false
FY
0001563665
No
No
Yes
Yes
0.5
0001563665
2023-01-01
2023-12-31
0001563665
2023-06-30
0001563665
2024-03-18
0001563665
2023-12-31
0001563665
2022-12-31
0001563665
2022-01-01
2022-12-31
0001563665
us-gaap:PreferredStockMember
HRGN:SeriesEConvertiblePreferredStockMember
2021-12-31
0001563665
us-gaap:CommonStockMember
2021-12-31
0001563665
us-gaap:AdditionalPaidInCapitalMember
2021-12-31
0001563665
us-gaap:RetainedEarningsMember
2021-12-31
0001563665
2021-12-31
0001563665
us-gaap:PreferredStockMember
HRGN:SeriesEConvertiblePreferredStockMember
2022-12-31
0001563665
us-gaap:CommonStockMember
2022-12-31
0001563665
us-gaap:AdditionalPaidInCapitalMember
2022-12-31
0001563665
us-gaap:RetainedEarningsMember
2022-12-31
0001563665
us-gaap:PreferredStockMember
HRGN:SeriesEConvertiblePreferredStockMember
2022-01-01
2022-12-31
0001563665
us-gaap:CommonStockMember
2022-01-01
2022-12-31
0001563665
us-gaap:AdditionalPaidInCapitalMember
2022-01-01
2022-12-31
0001563665
us-gaap:RetainedEarningsMember
2022-01-01
2022-12-31
0001563665
us-gaap:PreferredStockMember
HRGN:SeriesEConvertiblePreferredStockMember
2023-01-01
2023-12-31
0001563665
us-gaap:CommonStockMember
2023-01-01
2023-12-31
0001563665
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-12-31
0001563665
us-gaap:RetainedEarningsMember
2023-01-01
2023-12-31
0001563665
us-gaap:PreferredStockMember
HRGN:SeriesEConvertiblePreferredStockMember
2023-12-31
0001563665
us-gaap:CommonStockMember
2023-12-31
0001563665
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001563665
us-gaap:RetainedEarningsMember
2023-12-31
0001563665
HRGN:HarvardBiosciencePlanMember
2013-10-31
2013-10-31
0001563665
us-gaap:MoneyMarketFundsMember
2023-12-31
0001563665
us-gaap:RestrictedStockUnitsRSUMember
2023-01-01
2023-12-31
0001563665
HRGN:ComputerEquipmentAndSoftwareMember
2023-12-31
0001563665
srt:MinimumMember
HRGN:FurnitureMachineryAndEquipmentMember
2023-12-31
0001563665
srt:MaximumMember
HRGN:FurnitureMachineryAndEquipmentMember
2023-12-31
0001563665
us-gaap:LeaseholdImprovementsMember
2023-12-31
0001563665
us-gaap:LeaseholdImprovementsMember
2022-12-31
0001563665
us-gaap:MachineryAndEquipmentMember
2023-12-31
0001563665
us-gaap:MachineryAndEquipmentMember
2022-12-31
0001563665
us-gaap:ComputerEquipmentMember
2023-12-31
0001563665
us-gaap:ComputerEquipmentMember
2022-12-31
0001563665
2017-12-31
0001563665
2022-02-28
0001563665
HRGN:MedmarcMember
2022-03-03
2022-03-03
0001563665
HRGN:HarvardBioscienceInc.Member
2022-01-01
2022-06-30
0001563665
HRGN:HarvardBioscienceInc.Member
2023-01-01
2023-12-31
0001563665
HRGN:HarvardBioscienceInc.Member
us-gaap:ConvertiblePreferredStockMember
2023-01-01
2023-12-31
0001563665
HRGN:HarvardBioscienceInc.Member
HRGN:SeriesEEightPercentageConvertiblePreferredStockMember
2022-06-10
2022-06-10
0001563665
HRGN:HarvardBioscienceInc.Member
HRGN:SeriesEEightPercentageConvertiblePreferredStockMember
2022-06-10
0001563665
HRGN:YaleUniversityMember
2023-12-31
0001563665
HRGN:McGowanInstituteForRegenerativeMedicineMember
2023-12-31
0001563665
srt:MinimumMember
2023-12-31
0001563665
srt:MaximumMember
2023-12-31
0001563665
us-gaap:ResearchAndDevelopmentExpenseMember
2023-01-01
2023-12-31
0001563665
us-gaap:ResearchAndDevelopmentExpenseMember
2022-01-01
2022-12-31
0001563665
us-gaap:SellingAndMarketingExpenseMember
2023-01-01
2023-12-31
0001563665
us-gaap:SellingAndMarketingExpenseMember
2022-01-01
2022-12-31
0001563665
us-gaap:SellingGeneralAndAdministrativeExpensesMember
2023-01-01
2023-12-31
0001563665
us-gaap:SellingGeneralAndAdministrativeExpensesMember
2022-01-01
2022-12-31
0001563665
us-gaap:DomesticCountryMember
2023-12-31
0001563665
us-gaap:StateAndLocalJurisdictionMember
2023-12-31
0001563665
HRGN:HarvardBioscienceIncMember
HRGN:SeriesEConvertiblePreferredStockMember
2022-04-28
2022-04-28
0001563665
HRGN:HarvardBioscienceIncMember
HRGN:SeriesEConvertiblePreferredStockMember
2022-06-10
2022-06-10
0001563665
HRGN:SeriesEConvertiblePreferredStockMember
2023-01-17
2023-01-18
0001563665
us-gaap:CommonStockMember
2023-01-17
2023-01-18
0001563665
us-gaap:PrivatePlacementMember
2023-04-12
2023-04-12
0001563665
us-gaap:PrivatePlacementMember
us-gaap:SeriesEPreferredStockMember
2023-04-12
2023-04-12
0001563665
us-gaap:SeriesEPreferredStockMember
2023-04-12
0001563665
us-gaap:SeriesEPreferredStockMember
2023-04-12
2023-04-12
0001563665
HRGN:UndesignatedPreferredStockMember
2023-12-31
0001563665
us-gaap:SeriesBPreferredStockMember
2023-12-31
0001563665
us-gaap:SeriesCPreferredStockMember
2023-12-31
0001563665
us-gaap:SeriesDPreferredStockMember
2023-12-31
0001563665
us-gaap:SeriesEPreferredStockMember
2023-12-31
0001563665
us-gaap:CommonStockMember
us-gaap:PrivatePlacementMember
2023-04-12
2023-04-12
0001563665
us-gaap:PrivatePlacementMember
2023-04-12
0001563665
us-gaap:CommonStockMember
us-gaap:PrivatePlacementMember
2022-05-12
2022-05-12
0001563665
us-gaap:CommonStockMember
us-gaap:PrivatePlacementMember
2022-05-12
0001563665
us-gaap:PrivatePlacementMember
2022-05-12
2022-05-12
0001563665
us-gaap:PrivatePlacementMember
2022-05-12
0001563665
us-gaap:PrivatePlacementMember
2022-05-12
2022-05-16
0001563665
us-gaap:CommonStockMember
us-gaap:PrivatePlacementMember
2022-05-12
2022-05-16
0001563665
us-gaap:WarrantMember
us-gaap:PrivatePlacementMember
2022-05-12
2022-05-16
0001563665
HRGN:HarvardBioscienceInc.Member
HRGN:SeriesEConvertiblePreferredStockMember
2022-06-01
2022-06-30
0001563665
HRGN:HarvardBioscienceInc.Member
HRGN:SeriesEConvertiblePreferredStockMember
2022-01-01
2022-12-31
0001563665
HRGN:EmployeeStockPurchasePlanMember
2023-01-01
2023-12-31
0001563665
HRGN:EmployeeStockPurchasePlanMember
2023-12-31
0001563665
HRGN:EmployeeStockPurchasePlanMember
2022-12-31
0001563665
2022-05-12
2022-05-12
0001563665
2022-05-12
0001563665
us-gaap:WarrantMember
2021-12-31
0001563665
us-gaap:WarrantMember
2022-01-01
2022-12-31
0001563665
us-gaap:WarrantMember
2022-12-31
0001563665
us-gaap:WarrantMember
2023-12-31
0001563665
HRGN:HRGNAmendedAndRestateEquityIncentivePlanMember
2023-01-01
2023-12-31
0001563665
HRGN:HRGNAmendedAndRestateEquityIncentivePlanMember
2023-12-31
0001563665
us-gaap:PerformanceSharesMember
2023-12-31
0001563665
HRGN:HRGNAmendedAndRestateEquityIncentivePlanMember
2023-12-29
0001563665
us-gaap:ResearchAndDevelopmentExpenseMember
HRGN:Biostage2013EquityIncentivePlanMember
2023-01-01
2023-12-31
0001563665
us-gaap:ResearchAndDevelopmentExpenseMember
HRGN:Biostage2013EquityIncentivePlanMember
2022-01-01
2022-12-31
0001563665
us-gaap:SellingGeneralAndAdministrativeExpensesMember
HRGN:Biostage2013EquityIncentivePlanMember
2023-01-01
2023-12-31
0001563665
us-gaap:SellingGeneralAndAdministrativeExpensesMember
HRGN:Biostage2013EquityIncentivePlanMember
2022-01-01
2022-12-31
0001563665
HRGN:Biostage2013EquityIncentivePlanMember
2023-01-01
2023-12-31
0001563665
HRGN:Biostage2013EquityIncentivePlanMember
2022-01-01
2022-12-31
0001563665
us-gaap:WarrantMember
2023-01-01
2023-12-31
0001563665
us-gaap:WarrantMember
2022-01-01
2022-12-31
0001563665
us-gaap:EmployeeStockOptionMember
2023-01-01
2023-12-31
0001563665
us-gaap:EmployeeStockOptionMember
2022-01-01
2022-12-31
0001563665
HRGN:SeriesEConvertiblePreferredStockMember
2023-01-01
2023-12-31
0001563665
HRGN:RegenerativeBiotechMember
2023-01-01
2023-12-31
0001563665
HRGN:LongevityProductsMember
2023-01-01
2023-12-31
0001563665
HRGN:RegenerativeBiotechMember
2023-12-31
0001563665
HRGN:LongevityProductsMember
2023-12-31
0001563665
us-gaap:SubsequentEventMember
2024-03-31
0001563665
us-gaap:SubsequentEventMember
srt:ChiefExecutiveOfficerMember
2024-02-01
0001563665
us-gaap:SubsequentEventMember
2024-02-01
iso4217:USD
xbrli:shares
iso4217:USD
xbrli:shares
xbrli:pure
HRGN:Segment
HRGN:Item
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
10-K
| ☒ |
Annual
report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
| |
|
| For
the fiscal year ended December 31, 2023 |
| or |
| |
|
| ☐ |
Transition
report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For
the transition period from to
Commission
File Number 001-35853
Harvard
Apparatus Regenerative Technology, Inc.
(Exact
Name of Registrant as Specified in Its Charter)
| Delaware |
|
45-5210462 |
| (State
or other jurisdiction of |
|
(I.R.S.
Employer |
| Incorporation
or organization) |
|
Identification
No.) |
84
October Hill Road, Suite 11, Holliston, Massachusetts 01746
(Address
of Principal Executive Offices, including zip code)
(774)
233-7300
(Registrant’s
telephone number, including area code)
Securities
registered pursuant to Section 12(b) of the Act: None
| Title
of each class |
|
Trading
Symbol(s) |
|
Name
of each exchange on which registered |
| |
|
|
|
|
Securities
registered pursuant to Section 12(g) of the Act:
Common
Stock, $0.01 par value
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
YES
☐ NO ☒
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
YES
☐ NO ☒
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
YES
☒ NO ☐
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files).
YES
☒ NO ☐
Indicate
by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting
company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,”
“smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large
accelerated filer ☐ |
Accelerated
filer ☐ |
| Non-accelerated
filer ☒ |
Smaller
reporting company ☒ |
| |
Emerging
growth company ☐ |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report. ☐
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate
by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation
received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b).
☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act.
YES
☐ NO ☒
The
aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, computed by reference to
the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of June 30, 2023 was
approximately $33.8 million based on the closing sale price on that date of $3.65. Shares of the registrant’s common stock held
by each officer and director and each person known to the registrant to own 10% or more of the outstanding voting power of the registrant
have been excluded in that such persons may be deemed affiliates. This determination of affiliate status is not a determination for other
purposes.
At
March 18, 2024, there were 13,947,324 shares of the registrant’s common stock issued and outstanding.
DOCUMENTS
INCORPORATED BY REFERENCE
Portions
of the Company’s definitive Proxy Statement in connection with the 2024 Annual Meeting of Stockholders, to be filed within 120
days after the end of the Registrant’s fiscal year, are incorporated by reference into Part III of this Form 10-K. Except with
respect to information specifically incorporated by reference in this Form 10-K, such Proxy Statement is not deemed to be filed as part
hereof.
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC.
TABLE
OF CONTENTS
ANNUAL
REPORT ON FORM 10-K
For
the Year Ended December 31, 2023
INDEX
Forward-Looking
Statements
This
Annual Report on Form 10-K contains statements that are not statements of historical fact and are forward-looking statements within the
meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934 (the Exchange Act), each
as amended. The forward-looking statements are principally, but not exclusively, contained in “Item 1: Business” and “Item
7: Management’s Discussion and Analysis of Financial Condition and Results of Operations.” These statements involve known
and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different
from any future results, performance or achievements expressed or implied by the forward-looking statements. Forward-looking statements
include, but are not limited to, statements about management’s confidence or expectations and our plans, objectives, expectations
and intentions that are not historical facts. In some cases, you
can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,”
“would,” “seek,” “expect,” “plans,” “aim,” “anticipates,” “believes,”
“estimates,” “projects,” “predicts,” “intends,” “think,” “continue,”
“potential,” “is likely,” “permit,” “objectives,” “optimistic,” “new,”
“goal,” “target,” “strategy” and similar expressions intended to identify forward-looking statements.
These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties.
Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss many of these risks in
detail under the heading “Item 1A. Risk Factors” beginning on page 24 of this Annual Report on Form 10-K. You should carefully
review all of these factors, as well as other risks described in our public filings, and you should be aware that there may be other
factors, including factors of which we are not currently aware, that could cause these differences. Also, these forward-looking statements
represent our estimates and assumptions only as of the date of this Annual Report. We may not update these forward-looking statements,
even though our situation may change in the future, unless we have obligations under the federal securities laws to update and disclose
material developments related to previously disclosed information. Harvard Apparatus Regenerative Technology, Inc. is referred to herein
as “we,” “our,” “us,” and “the Company.”
PART
I
Item
1. Business.
OVERVIEW
Regenerative Biotech
We
are a clinical-stage biotechnology company focused on the development of regenerative medicine treatments for disorders of the gastro-intestinal
system and other organs that result from cancer, trauma or birth defects. Our technology is based on our proprietary cell-therapy platform
that uses a patient’s own stem cells to regenerate and restore function to damaged organs. We believe that our technology represents
a next generation solution for restoring organ function because it allows the patient to regenerate their own organ, thus eliminating
the need for human donor or animal transplants, the sacrificing of another of the patient’s own organs or permanent artificial
implants.
Our
technology uses mesenchymal stem cells that are retrieved via biopsy from the patient’s abdominal adipose, or fat, tissue prior
to surgery. These stem cells are isolated, expanded and then cultured on a tubular scaffold made from extremely thin fibers of polyurethane.
The scaffold is then incubated in a customized bioreactor where the stem cells continue to grow and adhere to the fibers of the scaffold.
The finished graft is then surgically implanted to replace the resected portion of the damaged organ. Several weeks after surgery, once
a conduit has been established, the implanted scaffold is removed using an endoscope. No permanent artificial implant remains in the
body.
We
conducted the world’s first successful regeneration of the esophagus in a cancer patient in August 2017. This surgery was
performed by Dr. Dennis Wigle, Chair of Thoracic Surgery at the Mayo Clinic in a patient requiring reconstruction of his esophagus
following the removal of a large tumor in his chest. The results were published in the Journal of Thoracic Oncology Clinical and
Research Reports in August 2021. The procedure demonstrated that using Harvard Apparatus Regenerative Technology, Inc.’s, or
HRGN’s, technology, we were able to successfully regenerate esophageal tissue, including the mucosal lining, to restore the
integrity, continuity and functionality of the esophageal tube.
Based on our successful first-in-human procedure and our preclinical
procedures in over 50 pigs, the U.S. Food and Drug Administration, or FDA has approved our Investigational New Drug (IND) application
to begin a phase 1 clinical trial for esophageal regeneration. This open-label trial will assess both safety and efficacy in up to ten
patients requiring esophageal reconstruction for any reason, including caustic burns, puncture wounds or damage to the esophagus following
chemoradiation therapy for esophageal cancer, at up to five U.S. hospitals. We have contracted with IQVIA, a leading global provider of
advanced analytics, technology solutions and clinical research services to the life sciences industry, as the contract research organization
(CRO) to manage our first clinical trial. We activated two clinical trial sites, the Mayo Clinic and the University of Michigan Medical
Center, and started screening patients in the third quarter of 2023. We continue to seek our first eligible patient for enrollment in
the trial.
We are initially targeting regeneration of the organs of the gastro-intestinal
tract and the airway, where organ transplants are not medically possible today. Human-donor organ transplants or animal xenotransplants
are currently not performed for these organs due to high rates of rejection. Additionally, we believe that our technology and intellectual
property will allow us to develop organ-regeneration treatments for other organs.
Our
product candidates are currently in development and have not yet received regulatory approval for sale anywhere in the world.
Longevity Products
In
the second quarter of 2023, our subsidiary in Hong Kong, Harvard Apparatus Regenerative Technology Limited, or Longevity Products, started focusing
on sales of longevity products.
Longevity Products plans to include a
broad range of products focused on personal healthcare including longevity dietary supplements. The Company started selling longevity
supplements through Longevity Products in the third quarter of 2023. These products are marketed to the general public and initially targeted at
consumers in the Great China Region through eCommerce (online sales).
Our
Pipeline
We believe our organ-regeneration technology has the potential for
broad applications in the field of medicine, for the repair or replacement of diseased or damaged organs. We are initially targeting conditions
of the esophagus, including traumatic injury, caustic burns, tissue damage following chemoradiation therapy and birth defects. Additional
product candidates in our development pipeline are targeted at the reconstruction of the colon and uterus wound repair.

Our
Strategy
Our
strategy is to develop and advance our pipeline of products, beginning with our lead product for the treatment of esophageal cancer,
through clinical development and commercialization. The key elements of our strategy include:
| |
● |
Initiate
the phase 1 clinical trial for our lead product candidate, the CellspanTM Esophageal Implant (CEI), for the treatment
of severe esophageal disease. Based upon our successful initial case of esophageal regeneration and our animal models, the FDA has
approved our IND application to commence a clinical trial in up to ten patients. We activated two clinical trial sites, the Mayo
Clinic and the University of Michigan Medical Center, and started screening patients in the third quarter of 2023. We continue to
seek our first eligible patient for enrollment in the trial. |
| |
|
|
| |
● |
Advance
our other pipeline products through clinical development. Based on the establishment of a favorable safety and efficacy profile that
we expect to demonstrate in our phase 1 clinical trial for regeneration of the esophagus, we intend to initiate a clinical trial
for the treatment of esophageal atresia, a rare birth defect where the esophagus does not fully develop, and the affected infant
is unable to swallow food. As we build our safety and efficacy data, we plan to initiate clinical trials in other areas including
injury, birth defects and diseases in the colon and other tubular organs that require reconstruction. |
| |
|
|
| |
● |
Develop
our technology for use in other life-threatening conditions that have a relatively short time to market. We believe our technology
has broad applications to treat organ failure. We intend to develop products focused on life-threatening conditions where current
treatments are ineffective, expensive or both. Many organ failures are orphan diseases, and we have orphan drug designations from
the FDA on our product candidates for severe disease in the esophagus. We believe that developing products for such conditions will
require smaller clinical trials and an overall less expensive development pathway than developing treatments for less severe conditions. |
| |
|
|
| |
● |
Pursue
development pathways in international markets. In addition to the U.S., we intend to pursue regulatory approval for our products
in several key international markets, including China, Europe and the U.K. Many of the conditions we are targeting have significantly
higher patient populations in foreign countries than in the U.S., thereby making them attractive commercial markets. We intend to
engage foreign health regulatory bodies to develop clinical and regulatory strategies to gain international approvals. In addition,
we have received Orphan Drug Designation from the European Medicines Agency (EMA) for the use of our CEI for esophageal atresia. |
| |
|
|
| |
● |
Collaborate
with leading medical and research institutions to develop our products and build awareness. We intend to continue to collaborate
with thought-leading medical institutions as we continue clinical development of our products and ultimately reach commercialization.
We currently have a co-development initiative with the Mayo Clinic, Connecticut Children’s Medical Center, Yale University
and the McGowan Institute for Regenerative Medicine at the University of Pittsburgh. We intend to build additional partnerships and
collaborations with leading institutions that we believe will help to drive awareness of our products and increase the likelihood
of market adoption. |
The
Problem
According to the American Cancer Society, every year approximately
17,000 Americans are diagnosed with esophageal cancer and approximately 15,000 of these diagnosed patients die from it. A year after being
diagnosed with esophageal cancer, 50% of patients die. After five years, 80% of these patients die. According to the World Health Organization’s
International Agency for Research on Cancer, every year, there are more than 600,000 patients diagnosed with esophageal cancer worldwide.
A current treatment option for patients with esophageal cancer is to
receive neoadjuvant therapy which can include definitive chemoradiation treatment. In many cases definitive chemoradiation treatment causes
damage to the esophagus leading to severe strictures (constrictions that close the throat and prevent swallowing) or fistulas (holes in
the tissue). The current treatment for the removal of the diseased part of the esophagus following chemoradiation is to surgically remove
the entire esophagus in a surgical procedure called an esophagectomy. The gap left by the removal of the diseased part of the esophagus
is then repaired using one of two difficult and expensive surgeries, both of which have frequent and significant complications. The first
type of surgery is gastric pull-up. In this surgery, the patient’s stomach is reshaped into a tube and pulled up from the abdomen
into the chest to connect to the top of the esophagus. With gastric pull-up, the patient no longer has a stomach with which to digest
food. In the second type of surgery, termed colonic interposition, a piece of the patient’s bowel is cut out and used to bridge
the gap where the diseased esophagus was removed. With colonic interposition, the patient often has insufficient intestine to digest food
properly. Both surgical procedures have high rates of complications such as damage to the lungs and infections caused by leakage of stomach
acids into the chest. Even with these surgical treatments, esophageal cancer is one of the deadliest forms of cancer.
In
addition to cancer, there are other injuries to the esophagus such as fistulas (holes), injuries caused by the accidental ingestion of
acids and alkalis, and birth defects. These are all difficult to treat surgically and often have significant long-term complications.
Hence,
there is an enormous need for, and a huge market for, a better treatment for cancer, injuries, and birth defects of the esophagus.
Our
Solution –Organ-Regeneration Technology
Our organ-regeneration technology uses a patient’s own stem cells
seeded on a temporary scaffold to regrow and restore their damaged organ. We believe our technology has numerous advantages over other
attempts to restore organ function because our implant is not a transplant of a human-donor or animal organ. It is also not a piece of
one of the patient’s other organs, and it is not an artificial implant that remains permanently in the body. Rather, our implants
will allow the patient to regenerate their own organ inside their own body.
Our
esophageal implant consists of a hollow, tubular scaffold consisting of a thin polyurethane fiber mesh that is formed in the shape of
the damaged section of the organ. This scaffold is then placed into a customized bioreactor and seeded with the patient’s own mesenchymal
stem cells which are obtained a few weeks before surgery through a biopsy of adipose (fat) tissue from the patient’s abdomen. The
stem cells are isolated and expanded and then seeded onto the tubular scaffold for incubation and further cell expansion. During several
days of incubation in our bioreactor, the stem cells attach to and grow into the outer 25% of the scaffold. The stem cell-seeded scaffold
is then surgically implanted into the patient to bridge the gap created where the diseased or damaged part of the esophagus was removed.
The
stem cells then stimulate the body’s natural wound-healing process including stimulating new blood vessel formation, scar-tissue
formation and the remodeling of that scar tissue into esophageal tissue. The scaffold guides the growth of new cells to regenerate the
esophagus. After approximately one month, a complete biological tube, or conduit, has formed and after approximately three months, the
tube has developed into a layered structure that contains the critical blood supply, muscles, and mucous-secreting glands to create a
functioning esophagus. At this point, the implanted scaffold is removed, as it is not a permanent implant.
Our
Technology Platform: How the Esophageal Implant Works
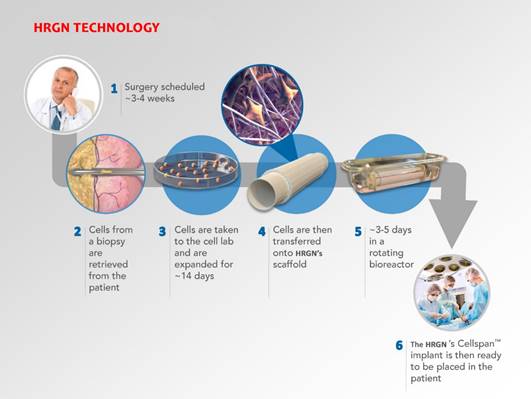
The
bioreactor and scaffold are made in our clean-room facilities in Holliston, Massachusetts and the cell seeding is performed at the FDA-approved,
clinical-grade human cell culture facility at the University of Texas Medical Branch.
Our
manufacturing process for the bioreactors and scaffolds has been approved by the FDA for the clinical trial. Based on expected FDA inspections,
additional development may be necessary for product approval.
For
our scaffolds, our primary materials are medical-grade plastic resins and solvents used to liquefy the resins in our manufacturing process.
These materials are readily available from a variety of suppliers and do not currently represent a large proportion of our total costs.
For our automatic cell seeding device and bioreactors, we perform final assembly and testing of components that we buy from third parties
like machine shops, parts distributors, molding facilities and printed circuit board manufacturers. These manufacturing operations are
performed primarily at our Holliston, Massachusetts headquarters.
Advantages
of the Esophageal Implant
Compared
with the current standard of care procedures for esophageal cancer patients - gastric pull-up or colonic interposition, our esophageal
implant offers the following major advantages:
| |
● |
Patients
can avoid the frequently life-threatening complications of either gastric pull up or colonic interposition surgery; |
| |
● |
Autologous
stem cells eliminate the risk of immune system rejection; |
| |
● |
The
procedure does not require the sacrifice of the patient’s stomach or colon, so those organs remain intact and function accordingly; |
| |
● |
It
leaves no permanent implant or artificial structure in the body. Permanent implants can lead to long-term complications, including
infection, which can lead to further surgical procedures including removal; |
| |
● |
Patients
can remain on a reasonable diet after a procedure with our esophageal implant. |
We
believe that these significant medical advantages will lead to strong demand from patients and doctors for our esophageal implant. Additionally,
we believe that it will receive a favorable reimbursement profile from payors and insurance companies because of the high cost and complications
associated with alternative procedures.
First-In-Human
Use of the Esophageal Implant and Scientific Proof of Esophageal Regeneration
On August 7, 2017, we announced the use of our esophageal implant in
a patient at the Mayo Clinic via an FDA-approved single-use expanded access, or compassionate use, application. The patient was a 75-year-old
male with a life-threatening cancerous mass in his chest that spanned his heart, a lung, and his esophagus. The surgery was performed
by Dr. Dennis Wigle, Chair of Thoracic Surgery. In order to remove the tumor, a portion of the heart was removed and repaired with a pericardial
patch, a portion of the lung was removed, a portion of the vena cava was repaired with a Gortex vascular graft, and a segment of the esophagus
was removed and repaired with our CEI. The patient’s surgeon informed us at that time that the surgery was successful, and the patient
was discharged from the hospital 42 days after implantation. The scaffold and stent were removed on day 104 after implantation.
In
February 2018, the surgeon informed us that the patient had died after living approximately eight months after surgery. The surgeon stated
that the cause of death was due to a stroke, and that the stroke was unrelated to the esophageal implant. The surgeon also informed us
that a preliminary autopsy had shown that the esophageal implant resulted in a regenerated esophageal tube in the patient, except for
a very small (approximately 5mm) hole outside the implant zone. The small hole was believed to be caused by abrasion from the Gortex
graft used to repair the vena cava. The surgeon also informed us that the esophageal regeneration in this patient was consistent with
the regeneration previously observed in our pig studies.
The
results were published in the Journal of Thoracic Oncology Clinical and Research Reports in August 2021. The photographs below, taken
from the paper, show the explanted esophagus from this procedure. The image on the left is the actual esophagus. The results from this
study, in combination with previously published results of esophageal regeneration using the CEI tissue-engineered graft 1,2
confirms that the regeneration process is reproducible in humans. In addition, the data presented confirms that epithelial
regeneration occurs during the initial wave of tissue regeneration and is typically complete by 3 months post-implantation.

1
La Francesca S, et al (2018). Long-term regeneration and remodeling of the pig esophagus after circumferential resection using
a retrievable synthetic scaffold carrying autologous cells. Sci Rep.; 8:4123.
2
Sundaram, S, et. al. (2022). Esophageal Regeneration following Surgical Implantation of a Tissue Engineered Esophageal Implant
in a Pediatric Model. NPJ Regen Med 7:1.
The
dark-brown tube in the center of the esophagus is the stent that was added to avoid narrowing of the esophagus. The stent for this patient
was changed twice, once prior to our esophageal implant scaffold removal and once after the scaffold and the second stent were removed.
The final stent was removed at five and a half months post-surgery. We anticipate that patients treated with our esophageal implant are
likely to undergo at least one stent exchange during their recovery with the discontinuation of stents by six to nine months post-surgery.
Stents are deployed and retrieved endoscopically, that is, via the mouth, and accordingly, there is no surgical incision in the chest.
The
images on the righthand side are photographs taken under a microscope to show the characteristics of the newly formed tissue extending
from the native tissue through a transition zone where the regeneration of the muscle layer begins and then into the center of the implant.
The native tissue has multiple layers composed of different cell types, including the mucosal layer on the lumenal surface, a submucosa
composed of connective tissue and smooth muscle cells and the muscular adventitia composed of smooth and striated muscle (brown staining
structures in the right most actin, α-SMA). In the right most panels, the brown coloration along the left side of the images shows a continuous
line of muscles running up the regenerated esophagus (arrows). These muscles are the muscularis mucosae which contract to eject mucous
into the esophagus. This mucosal lining is essential to the long-term survival of the patient because it both lubricates the esophagus
to allow food to be swallowed and provides a barrier to infection. This mucosal lining was seen at three months in both the human patient
and in our pig models.
In
this patient we saw the development of a tube comprised of the patient’s own tissue within one month, and the development of the
mucosal lining within three months. In pig models we have observed a similar regeneration process with the development of a tube within
one month and the development of the mucosal lining within three months. In our clinical trial, the primary endpoint is the development
of the tube of the patient’s tissue within three months and one of the secondary endpoints is the development of the mucosal lining
within twelve months.
Preclinical
Models - Pig Studies
The
pre-clinical animal studies using our esophageal implant investigated several key aspects of the product pertaining to the implant procedure,
cell survival, the architecture of the regenerated tissue at multiple survival time points, the post-implantation clinical management
procedures including Computed Tomography, or CT imaging to assess the growth of new tissue, esophageal stent management, endoscopy procedures,
barium swallow tests and nutritional management.
Following
implantation, CT imaging revealed early tissue deposition and the formation of a contiguous tissue conduit. Endoscopic evaluation at
multiple time points revealed complete epithelialization of the lumenal surface by day 90. Histologic evaluation at several necropsy
time points, post-implantation, demonstrated that the tissue continues to remodel over the course of a one-year survival time period,
resulting in the development of esophageal structural features, including the mucosal epithelium, muscularis mucosae, lamina propria,
as well as smooth muscle proliferation/migration initiating the formation of a laminated adventitia. One-year survival demonstrated restoration
of oral nutrition, normal animal growth and the overall safety of this treatment regimen.
The
image below is taken from a paper we published in Nature Partner Journals Regenerative Medicine in January 2022, in conjunction with
our development partner, Connecticut Children’s Medical Center.
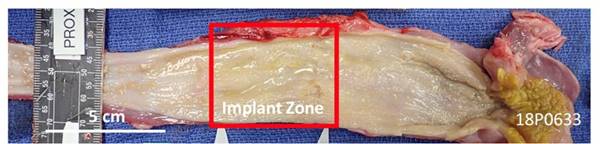
This
image shows an esophagus explanted from a pig 90 days after our esophageal implant was implanted. The implant zone is visually almost
identical to the native tissue to the left and right of it. We note the regeneration of the interior surface of the esophagus and
the regeneration of the surrounding tissue that is visible in red at the top of the red box. The red color of the surrounding tissue
indicates the presence of a healthy blood supply. We note further the glossy, reflective coating on the inside of the esophagus. This
is evidence of the mucosal lining which is essential to the long-term survival of the patient. This mucosal lining was seen at three
months in the pigs and was also observed in the human patient. The investigators concluded that at one year it was difficult to distinguish
neo-tissue versus the native tissue.
Current
Phase 1 Clinical Trial
Based on both the successful in-human procedure at the Mayo Clinic
and our extensive large animal research, the FDA has approved our Investigational New Drug application to commence our clinical trial.
The trial will be a ten-patient phase 1 trial, in up to five hospitals in the U.S. and is designed to measure both the safety and efficacy
of our product candidate in the patient population. Enrollment criteria includes any patient that requires removal of a part of the esophagus
that is up to six centimeters long for any medical reason. We expect enrolled patients to include esophageal cancer patients, post-neoadjuvant
chemoradiation therapy, but we may enroll patients with other esophageal conditions that require reconstruction and regeneration. We activated
two clinical trial sites, the Mayo Clinic and the University of Michigan Medical Center, and started screening patients in the third quarter
of 2023. We continue to seek our first eligible patient for enrollment in the trial.
The
primary endpoint of the upcoming trial is the establishment of a continuous biological neoconduit, or tube, by three months post-surgery.
We saw this tube at one month in the human patient and in the pigs. One of the secondary endpoints will be the development of a mucosal
lining in the esophagus by twelve months post-surgery. We saw this mucosal lining by three months in the human patient and the pigs.
Because we reached the primary endpoint and one of the secondary endpoints in both the human patient and the pigs, we believe that we
have a high likelihood of success in this clinical trial.
Based
on the FDA’s approval of our clinical trial for any condition that requires removal of part of the esophagus, we believe that we
are able to pursue the treatment of multiple diseases, injuries or birth defects with a single clinical trial. As a result, we believe
that this clinical trial will advance the Cellspan Esophageal Implant for numerous indications with the possibility of treating esophageal
damage due to cancer, Barrett’s esophagus, fistulas, traumatic and potentially long-term complications from birth defects in the
esophagus. Compared to developing treatments for a single underlying medical condition, we believe that addressing multiple medical conditions
in a single clinical trial has the potential to significantly reduce our costs to expand the market for our products.
Preclinical
Development
In
January 2022, together with Connecticut Children’s Medical Center, we published in Nature Partner Journals Regenerative Medicine
the results of implanting pediatric-sized esophageal implants in 15 piglets. Numerous survival times were histologically analyzed to
understand the tissue development and timing of the regeneration. Overall, the graft implantation procedure was deemed safe and feasible.
The piglets showed regeneration of a conduit, or tube, by one month and the regeneration of a normal mucosal lining by three months.
Additionally, histological evaluation demonstrated that the tissue continued to develop throughout the course of the one-year survival
period. Importantly, the piglets also showed normal growth and weight gain which are considered critical in treating human babies.
This
research also utilized post-surgical techniques that closely mimic the hospital care that human infants undergo to treat esophageal atresia.
These techniques included non-invasive CT imaging of the regenerated tissue, parenteral feeding via a G tube which are normally used
to feed human infants after surgeries in the gastro-intestinal tract as well as endoscopy using a pediatric endoscope.
Clinical
Pathway
We
believe that this study laid both the scientific and clinical groundwork for treating babies with birth defects in the esophagus with
the Cellspan Esophageal Implant. The FDA approval for the clinical trial allows us to treat children once we have established safety
in adult patients in the phase 1 clinical trial. Once we have established the safety of the implant in adults, we expect to recruit children
into a clinical trial for esophageal atresia.
Orphan
Drug Designation – Seven Years of Exclusivity
In
November 2016, we were granted Orphan Drug Designation for our esophageal implant by the FDA to restore the structure and function of
the esophagus subsequent to esophageal damage due to cancer, injury or congenital abnormalities. We also were granted Orphan Drug Designation
for trachea on September 4, 2014.
The
Orphan Drug Act provides incentives to manufacturers to develop and market drugs and biologics for rare diseases and conditions affecting
fewer than 200,000 persons in the U.S. at the time of application for orphan drug designation, or more than 200,000 individuals in the
U.S. and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in
the U.S. for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested
before submitting a new drug application, or NDA, or Biologics License Application or BLA. After the FDA grants orphan product designation,
the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does
not convey any advantage in or shorten the duration of the regulatory review and approval process. The first developer to receive FDA
marketing approval for an orphan biologic is entitled to a seven-year exclusive marketing period in the U.S. for that product as well
as a waiver of the BLA user fee. The exclusivity prevents FDA approval of another application for the same product for the same indication
for a period of seven years, except in limited circumstances where there is a change in formulation in the original product and the second
product has been proven to be clinically superior to the first. In addition, Orphan Drug Designation provides a seven-year marketing
exclusivity period against competition in the U.S. from the date of a product’s approval for marketing. This exclusivity would
be in addition to any exclusivity we may obtain from our patents. Additionally, orphan designation provides certain incentives, including
tax credits and a waiver of the BLA fee. We also plan to apply for Orphan Drug Designation for our esophageal implant in Europe. Orphan
Drug Designation in Europe would provide market exclusivity in Europe for a period of ten years from the date of the product’s
approval for marketing.
Our
Strategy
Our
strategy is to develop and advance our pipeline of products, beginning with our lead product for the treatment of esophageal cancer,
through clinical development and commercialization. The key elements of our strategy include:
| |
● |
Initiate
the phase 1 clinical trial for our lead esophageal implant product candidate for the treatment of severe esophageal disease. Based
upon our successful initial case of esophageal regeneration and our animal models, the FDA has approved our IND application to commence
a clinical trial in up to ten patients. We activated two clinical trial sites, the Mayo Clinic and the University of Michigan Medical
Center, and started screening patients in the third quarter of 2023. We continue to seek our first eligible patient for enrollment
in the trial. |
| |
|
|
| |
● |
Advance
our other pipeline products through clinical development. Based on the establishment of a favorable safety and efficacy profile
that we expect to demonstrate in our phase 1 clinical trial for regeneration of the esophagus, we intend to initiate a clinical trial
for the treatment of esophageal atresia, a rare birth defect. As we build our safety and efficacy data, we plan to initiate clinical
trials in other areas following the demonstration of efficacy in animal models, including colon resection and the prevention of intrauterine
adhesions. |
| |
|
|
| |
● |
Develop
our technology for use in other life-threatening conditions that have a relatively shorter time to market. We intend to develop
products focused on life-threatening conditions where current treatments are ineffective, expensive or both. Many organ failures
are orphan diseases, and we have orphan drug designations from the FDA on our product candidates for severe disease in both the esophagus
and the colon. We believe that developing products for such conditions will require smaller clinical trials and an overall less expensive
development pathway than developing treatments for less severe conditions. |
| |
|
|
| |
● |
Pursue
development pathways in international markets. In addition to the U.S., we intend to pursue regulatory approval for our products
in several key international markets, including China, Europe and the U.K. Many of the conditions we are targeting, have significantly
higher patient populations in foreign countries than in the U.S., thereby making them attractive commercial markets. We intend to
engage foreign health regulatory bodies to develop clinical and regulatory strategies to gain international approvals. |
| |
|
|
| |
● |
Collaborate
with leading medical and research institutions to develop our products and build awareness. We intend to continue to collaborate
with thought-leading medical institutions as we continue clinical development of our products and ultimately reach commercialization.
We currently have a co-development initiative with the Mayo Clinic and with the Connecticut Children’s Medical Center. We intend
to build additional partnerships and collaborations with leading institutions that we believe will help to drive awareness of our
products and increase the likelihood of market adoption. |
Our
Technology
Biocompatible
Scaffold Component
Our
proprietary biocompatible scaffold component of our esophageal implant is constructed primarily of extremely thin polyurethane fibers.
This material was chosen based on extensive testing of various materials. The scaffold is made using a manufacturing process known as
electrospinning. The combination of the electrospinning process, which provides control over the desired microstructure of the scaffold
fabric, with the polyurethane results in a scaffold that we believe has favorable biocompatibility characteristics.
The
Patient’s Cells
The
cells we seed onto the scaffold are obtained from the patient’s adipose tissue, or abdominal fat. This fat tissue is obtained from
a standard biopsy during the weeks leading up to the implant surgery. Mesenchymal stem cells are extracted and isolated from the adipose
tissue biopsy. The isolated cells are then expanded, or grown, for a short period prior to surgery in order to derive a sufficient cell
population to be seeded on the scaffold. The cells are then seeded on the scaffold in our proprietary bioreactor and incubated there
before the implant surgery.
Our
technology is protected by thirteen issued U.S. patents (including patents on the bioreactor, the structure of the scaffold and the retrievable
nature of the scaffold), two Orphan-Drug Designations from the FDA, both of which confer seven years of exclusivity in addition to protection
offered by the patents, and our first-mover advantage which allows us to improve the standard of care. Potential competitors would now
have to improve upon our new standard of care rather than just improve on the existing standard of care in order to get their product
candidates approved by the FDA. In addition, our patent claims cover patches as well as tubular structures. We intend to develop patches
for the repair of tubular organs as well as solid organs.
See
the “Intellectual Property, Licenses and Related Agreements” section below for more details.
Additional
Targeted Diseases
Targeted
Diseases
According
to the World Health Organization, or WHO, International Agency for Research on Cancer’s Global Cancer Observatory database, worldwide
there were over 600,000 cases of esophageal cancer in 2020. There are over one million cases of colon cancer. The following are the approximate
case counts by certain geographic region pertaining to the cancers noted below:
| | |
Case Count by Geography | |
| Cancer Type | |
USA | | |
China | | |
Japan | | |
Europe | | |
ROW | | |
Worldwide | |
| Esophagus Adults | |
| 18,309 | | |
| 324,422 | | |
| 26,262 | | |
| 52,993 | | |
| 182,114 | | |
| 604,100 | |
| Colon | |
| 101,809 | | |
| 306,078 | | |
| 96,781 | | |
| 325,335 | | |
| 318,512 | | |
| 1,148,515 | |
| Total | |
| 120,118 | | |
| 630,500 | | |
| 123,043 | | |
| 378,328 | | |
| 500,626 | | |
| 1,752,615 | |
Sources:
Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries Hyuna Sung,
PhD; Jacques Ferlay, MSc, ME; Rebecca L. Siegel, MPH; Mathieu Laversanne, MSc; Isabelle Soerjomataram, MD, MSc, PhD; Ahmedin Jemal, DMV,
PhD; Freddie Bray, BSc, MSc, PhD.
These
numbers of patients do not include those with fistulas, ulcers, injuries or birth defects, all of which we believe may be treatable with
our technology.
Because
our product candidates are likely to save or extend lives, improve the quality of life, and save money by reducing the complications
associated with current surgical repair techniques, we expect to charge more than $250,000 per product in the U.S. market.
Treating
even one tenth of only those patients who are diagnosed with esophageal cancer each year could generate billions of dollars in annual
revenue. We believe that the market potential for our products is significantly higher.
Esophageal
Disease
Esophageal
cancer is one of the deadliest types of cancer. According to the American Cancer Society, there are approximately 17,000 new diagnoses
of esophageal cancer in the U.S. each year, and there are more than 15,000 deaths. Typically, a year after diagnosis with esophageal
cancer, 50% of patients die and after 5 years, 80% die.
There
are approximately 600,000 new diagnoses of esophageal cancer globally each year, according to the World Health Organization’s International
Agency for Research on Cancer.
Hence,
there is a vast need for a better treatment for esophageal cancer.
Approximately
5,000 esophagectomy surgeries occur in the U.S. annually to treat esophageal cancer, and approximately 10,000 esophagectomies occur in
Europe annually. We believe that approximately one half of the world’s esophageal cancer cases occur in China, which would represent
the largest potential patient population for our adult esophageal product candidate. We believe that our esophageal implant, if approved,
has the potential to provide a major advance over the current esophagectomy procedures for addressing esophageal disease, which have
high complication and morbidity rates.
We
believe that our esophageal implant has the potential to provide physicians a new, simpler procedure to restore organ function while
significantly reducing complication and morbidity rates compared with the current standard of care, and without creating significant
quality of life issues for patients.
Pediatric
Esophageal Atresia
According
to the Centers for Disease Control and Prevention (CDC), each year it is estimated that approximately 4,100 children in the U.S. are
born with a congenital birth defect known as esophageal atresia. Esophageal atresia is a condition where an infant is born with an esophagus
that does not extend completely from the mouth to the stomach. When a long segment of the esophagus is lacking, the current standard
of care is a series of surgical procedures where sutures are applied to both ends of the esophagus in an attempt to stretch them and
pull them together so they can be surgically connected at a later date.
This
surgical process can take several weeks, and the procedure often involves serious complications and high rates of failure. The infant
usually must remain in the neonatal intensive-care unit for this time which can cost thousands of dollars per day. This process also
requires at least two separate surgical interventions. Other surgical options include the use of the child’s stomach or intestine
that would be pulled up into the chest to allow a connection to the mouth. These methods are similar to the use of gastric pull ups and
interpositioning used in adult patients and carry similar side effect and safety profiles. We are working in collaboration with the Connecticut
Children’s Medical Center, to advance an esophageal implant solution to address esophageal atresia that we believe will be more
effective, safer, and less expensive than the current procedures.
Colon
Cancer
Based
on input from our Scientific Advisory Board, which includes certain well-known surgeons in the field of regenerative medicine, we are
planning to research regenerating other parts of the gastro-intestinal tract such as the small intestine and colon. All these organs
require replacement when they are damaged by cancer, injury, and birth defects. There are over one million patients diagnosed with colon
cancer every year.
Infertility
Asherman’s Syndrome is a rare, acquired gynecologic condition
resulting from the buildup of scar tissue and intrauterine adhesions (IUA). IUA occur primarily after a dilation and curettage (D&C)
procedure for elective termination of pregnancy, a missed or incomplete miscarriage, or to treat a retained placenta after delivery. IUA
can go undetected, but in many cases can lead to altered menstrual cycles (hypomenorrhea) and can ultimately lead to infertility It is
estimated that up to 30% women of reproductive age (15-49 years) in the US that undergo elective pregnancy termination will develop IUA.
Based on the CDC recorded number of elective pregnancy termination of over 600,000 per year3, the incidence of Asherman’s
syndrome in the US is estimated to be in excess of 100,000 women in ages 15-49. We are currently evaluating our technology to prevent
IUA in an animal model of IUA with a prominent laboratory and Chief of Obstetrics and Gynecology.
3
Data and Statistics - Reproductive Health | CDC
Our
History
We
were incorporated under the laws of the State of Delaware on May 3, 2012 as a wholly-owned subsidiary of Harvard Bioscience, Inc., or
Harvard Bioscience, to provide a means for separating its regenerative medicine business from its other businesses. Harvard Bioscience
decided to separate its regenerative medicine business into our company, a separate corporate entity, or the Separation, and it spun
off its interest in our business to its stockholders in November 2013. Since the Separation we have been a separately traded public company
and Harvard Bioscience has not controlled our operations. Following the Separation, we continued to innovate our bioreactors based on
our physiology expertise, we developed our materials science capabilities and we investigated and developed a synthetic tracheal
scaffold. By that time, we had built and staffed cell biology laboratories at our Holliston facility, to give ourselves the ability to
perform and control our scientific investigation and developments internally. At that point, we began the second phase of our company’s
development.
In
mid-2014, we increased the pace of our scientifically based internal analysis and development of our first-generation tracheal implant
product candidate, the HART-Trachea . From large-animal studies conducted thereafter we found that the product candidate elicited
an unfavorable inflammatory response after implantation, which required additional development and testing. These requirements extended
our expectations regarding our regulatory milestones, and we announced the additional testing and extended milestone expectations in
January 2015. During 2015 we isolated and tested all major variables of the organ scaffold and the cell source and protocols, examining
the effects of alternatives against the then-existing product approach. Through extensive in vitro preclinical studies, and small-animal
and large-animal studies, we made dramatic improvements, and discovered that the mechanism of action of our current approach was very
different from our hypothesis regarding that of the first-generation product candidate. Our technology uses a different scaffold material
and microstructure, a different source and concentration of the patient’s cells and several other changes from our earlier trachea
initiative. These changes resulted in a scaffold that was temporary and could be removed via the mouth in an endoscopic procedure that
did not require major surgery in the chest. The temporary nature of the scaffold reduces the risk of long-term complications that can
arise from permanent implants such as those from hernia meshes and breast implants.
Clinical
Trials
The
FDA has approved our first clinical trial.
Based on both the successful human experience at the Mayo Clinic, and
our extensive large-animal research (we have performed surgeries on over 50 pigs including for both adult and pediatric diseases), the
FDA has approved our clinical trial. The trial will be a 10-patient phase 1 trial that measures both the safety and efficacy of our product
candidate in the patient population. This clinical trial is for any patient that requires removal of a part of the esophagus that is less
than 6cm long for any reason. The primary endpoint in the trial is the establishment of a continuous biological neo-conduit, or tube,
by three months. In the human patient, this tube was seen in one month. In our pig research, we have seen the formation of a conduit by
one month and sometimes by 14 days. One of the secondary endpoints will be the development of a mucosal lining in the esophagus by 12
months or earlier. In the one human patient treated so far, this mucosal lining was seen at three months. In our pig research, we have
seen this mucosal lining in three months.
Establishing
a safety profile in our current adult clinical trial will allow us to submit an IND for using our technology to treat esophageal atresia
in the pediatric population.
Our
esophageal implant will not be tested for safety on healthy volunteers (the usual goal of a phase 1 trial) or for dose-response and maximum-tolerated
dose (the usual goals of a phase 2 trial). Measuring safety and efficacy in the patient population is normally the goal of a phase 3
clinical trial. Hence, our approved trial is more similar to a small phase 3 clinical trial than a typical first clinical trial. We expect
to add patients to this clinical trial, including in Europe and China until we have sufficient data to gain approval.
Unlike
the normal drug discovery process, which assesses a drug for its ability to treat a single disease, we can pursue multiple diseases with
a single clinical trial. This is because any medical condition that requires the removal of part of the esophagus can be repaired with
our esophageal implant. It does not matter that the need to surgically remove part of the esophagus is caused by esophageal cancer, Barrett
esophagus (damage to the lower esophagus caused by the reflux of stomach acids into the esophagus), a fistula (a hole in the esophagus),
a birth defect, or a wound or injury to the esophagus. Our esophageal implant can be used to treat any of these conditions. Because of
this, we believe that the available market in treating the esophagus to be far larger than that for treating esophageal cancer alone.
In addition, we can access that large patient population without having to conduct a new clinical trial for each underlying medical condition.
Compared to the development of new drugs, this greatly reduces our costs to expand the market size for our products.
We
intend to request Fast Track status, Breakthrough Therapy designation, Regenerative Medicine Advanced Therapy, or RMAT, designation,
Accelerated Approval, Priority Review and a Priority Review Voucher from the FDA. There are many benefits of such designations, including
reduced costs and faster times to market. Please refer to the Regulatory Strategy section for more details.
Our
first clinical trial is in the U.S. for patients with cancer, injury, or birth defects in the esophagus. However, there are far more
patients with these conditions in Europe and Asia than there are in the U.S. For this reason, we intend to expand our clinical trial
to include patients in Europe and Asia and to seek regulatory approval in those countries as well.
In
addition to having large patient populations, for product candidates like ours, both the European Union, or E.U., and some countries
in Asia allow for “conditional approval”. Conditional approval is country specific but, in general, it would allow us to
market our products, and obtain revenue from the sales of the respective product, after successful phase 2 clinical trial results.
Conditional approval is granted subject to the regulatory authority being able to rescind the approval if something goes wrong as
more patients get treated. Hence, it is possible that we could see revenue in either Asia or the E.U. before we see revenue in the
U.S.
Research
and Development
Our
primary research and development activities are focused in three areas: materials science, cell biology and engineering. In
materials science, we focus on designing and testing biocompatible organ scaffolds, testing the structural integrity and the
cellularization capacities of the scaffolds. In cell biology, we focus on developing and testing isolation and expansion protocols,
cell characterization and cell fate studies, investigating the effects of various cell types and concentrations, evaluating the
biocompatibility of scaffolds, experimenting with different cell seeding methodologies, and developing protocols for implantation
experiments. Our engineering group supports the materials science and cell biology groups across an array of their activities, i.e.
designing, engineering and making our proprietary bioreactors and automatic cell seeding device. All three of our research and
development groups combine to plan and execute our in vitro studies. A fundamental part of our research and development
effort in developing our technology has been dedicated to the discovery and development of small and large-animal model
studies.
In
addition to our in-house engineering and scientific development team, we collaborate with leaders in the field of regenerative medicine
who are performing the fundamental research and surgeries in this field to develop and test new product candidates that will advance
and improve the procedures being performed. We will work with our collaborators to further enhance our product candidates to make them
more efficient and easier to use by surgeons. In the U.S., our principal collaborations have been with Mayo Clinic and Connecticut Children’s
Medical Center. Collaboration typically involves us developing new technologies specifically to address issues these researchers and
clinicians encounter, and then working together to translate our technology from pre-clinical studies to clinical trials. In certain
instances, we have entered into agreements that govern the ownership of the technologies developed in connection with these collaborations.
We
incurred approximately $3.1 million and $1.7 million of research and development expenses in 2023 and 2022, respectively. As we have
not yet applied for or received regulatory approval to market any clinical products, no amount of these research and development costs
have been passed on to our customers.
Manufacturing
and Resources
The
bioreactor and scaffold are made in our clean-room facilities in Holliston, Massachusetts and the cell seeding is currently performed
at the FDA-approved clinical-grade human cell culture facility at the University of Texas Medical Branch.
Our
manufacturing process for the bioreactors and scaffolds has been approved by the FDA for the clinical trial. Additional development is
likely to be necessary for product approval.
For
our scaffolds, our primary materials are medical-grade plastic resins and solvents used to liquefy the resins in our manufacturing process.
These materials are readily available from a variety of suppliers and do not currently represent a large proportion of our total costs.
For our automatic cell-seeding device and bioreactors, we perform final assembly and testing of components that we buy from third parties
like machine shops, parts distributors, molding facilities and printed circuit board manufacturers. These manufacturing operations are
performed primarily at our Holliston, Massachusetts headquarters.
Sales
and Marketing
We
expect that most surgeries using our esophageal implant will be performed at a relatively small number of major hospitals in the U.S.,
Asia and in Europe. In addition, our technology platform is initially aimed at treating the esophagus, the bronchi, and the trachea,
all of which are treated by thoracic surgeons. As a result, we expect to employ only a small sales force as compared to companies selling
treatments for larger patient populations.
We
expect to price the product commensurate with the medical value created for the patient and the costs avoided with the use of our product.
Because our products are likely to save or extend lives, improve the quality of life, and save money by reducing the complications associated
with current surgical repair techniques, we expect to charge approximately $250,000 per product in the U.S.
We
further expect to be paid by the hospital that buys the product from us. Finally, we expect that the hospital would seek reimbursement
from government payers, private health insurers and other third-party payers for the entire transplant procedure, including the use of
our products.
Intellectual
Property, Licenses, and Related Agreements
We have thirteen issued U.S. patents that cover the bioreactor, the
scaffold, and the surgical procedure. The patent claims cover the use of synthetic scaffolds for any use in the gastro-intestinal tract
and the airways. These patents include the claim of having a removable scaffold. The patent claims cover patches as well as tubes. We
intend to research the patch-based approach to treat damage to solid organs. We also have two issued patents in China, one patent issued
in Japan, two patents issued in Europe, two U.S. orphan-drug designations which can provide seven years of market exclusivity in the U.S.
market after market approval from the FDA and 1 EMA orphan drug designation, which can provide ten years of market exclusivity in the
European market after market approval from the EMA. There are numerous other filings pending. We expect these patents to provide protection
into the mid to late 2030’s.
Sublicense
Agreement with Harvard Bioscience
We
own the right to use the brand name “Harvard Apparatus Regenerative Technology” in the medical sciences field under a license
agreement with Harvard University via a sublicense from Harvard Bioscience. Harvard Bioscience’s right to use the name arises from
a license agreement, effective December 19, 2002, between it and the President and Fellows of Harvard University. Harvard Bioscience
began at Harvard University in 1903 as Harvard Apparatus and has a license to the name Harvard Apparatus in research and industrial fields.
Our right to use the name in the medical field arises from the sublicense signed when Harvard Apparatus Regenerative Technology was
separated from Harvard Bioscience in 2013 (as more fully described below). Harvard Bioscience delegated its right to use the name in
the medical field to us and Harvard Bioscience has no right to use the Harvard mark in the medical field. We intend to use this brand
name on our products in the future. We do not have the right to use the Harvard or Harvard Apparatus marks alone but only as Harvard
Apparatus Regenerative Technology. We believe we are the only licensee of the Harvard name in the medical products’ field. This
license is perpetual, worldwide and royalty-free. There are restrictions on our use of the name such as not using it in the color crimson
and not using it in a serifed font. We currently have no affiliation with Harvard University.
Separation
Agreements with Harvard Bioscience
On
November 1, 2013, to effect the Separation, Harvard Bioscience distributed all of the shares of our common stock to the Harvard Bioscience
stockholders, or the Distribution. Prior to the Distribution, Harvard Bioscience contributed the assets of its regenerative medicine
business, and approximately $15 million in cash, to our company to fund our operations following the Distribution.
In
connection with the Separation and immediately prior to the Distribution, we entered into a Separation and Distribution Agreement, Intellectual
Property Matters Agreement, Product Distribution Agreement, Tax Sharing Agreement, Transition Services Agreement, and Sublicense Agreement
with Harvard Bioscience to effect the Separation and Distribution and provide a framework for our relationship with Harvard Bioscience
after the Separation. These agreements govern the current relationships among us and Harvard Bioscience and provided for the allocation
among us and Harvard Bioscience of Harvard Bioscience’s assets, liabilities, and obligations (including employee benefits and tax-related
assets and liabilities) attributable to periods prior to the Separation.
Government
Regulation
Our
product candidates and our operations are subject to extensive regulation by the U.S. FDA and other federal and state authorities, as
well as comparable authorities in foreign jurisdictions, which are discussed below. The FDA is divided into various “Centers”
by product type such as the Center for Drug Evaluation and Research, or CDER, the Center for Biologics Evaluation and Research, or CBER,
and the Center for Devices and Radiological Health, or CDRH. Different Centers review drug, biologic, or device applications. Our product
candidates are subject to regulation as combination products, biologics and medical devices, in the United States under the Federal Food,
Drug, and Cosmetic Act, or FDCA, and the Public Health Services Act, or PHS Act, and their implementing regulations as implemented and
enforced by the FDA.
CBER
regulates medical devices related to licensed blood and cellular products by applying appropriate medical device laws and regulations.
Specifically, CBER regulates the medical devices involved in the collection, processing, testing, manufacture and administration of licensed
blood, blood components and cellular products. The medical devices regulated by CBER are intimately associated with the blood collection
and processing procedures as well as the cellular therapies regulated by CBER. CBER has developed specific expertise in blood, blood
products and cellular therapies and the integral association of certain medical devices with those biological products supports the regulation
of those devices by CBER. CBER also regulates biologics, which includes cells and tissues, serum, vaccines, blood and blood products,
and analogous substances.
After
receiving FDA approval or clearance, an approved or cleared product must comply with post-market safety reporting requirements applicable
to the product based on the application type under which it received marketing authorization. In the case of current good manufacturing
practices, or cGMP, the applicant may take one of two approaches: (1) complying with cGMP for each constituent part, or (2) a streamlined
approach specific to combination products, subject to certain limitations.
Regulatory
Strategy
Domestic
Regulation of our Product Candidates - FDA Approval Process
The
FDA extensively regulates, among other things, the research, development, testing, manufacture, quality control, approval, labeling,
packaging, storage, record-keeping, promotion, advertising, distribution, marketing and import and export of medical products. The FDA
governs the following activities that we may perform or that may be performed on our behalf, to ensure that the medical products we may
in the future manufacture, promote and distribute domestically or export internationally are safe and effective for their intended uses:
| |
● |
product
design, preclinical and clinical development and manufacture; |
| |
● |
product
premarket clearance and approval; |
| |
● |
product
safety, testing, labeling and storage; |
| |
● |
recordkeeping
procedures; |
| |
● |
product
marketing, sales and distribution; and |
| |
● |
post-marketing
surveillance, complaint handling and adverse event reporting, including reporting of deaths, serious injuries, malfunctions or other
deviations; and |
| |
● |
recall
of products, including repairs or remediation. |
The
labeling, advertising, promotion, marketing and distribution of biologics and medical devices also must be in compliance with the FDA
and U.S. Federal Trade Commission, or FTC, requirements which include, among others, standards and regulations for off-label promotion,
industry sponsored scientific and educational activities, promotional activities involving the internet, and direct-to-consumer advertising.
The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the
issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures,
injunctions and criminal prosecution. Recently, promotional activities for FDA-regulated products of other companies have been the subject
of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham
Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. In addition, we are required
to meet regulatory requirements in countries outside the U.S., which can change rapidly with relatively short notice.
The
FDA has broad post-market and regulatory enforcement powers. Manufacturers of biologics and medical devices are subject to unannounced
inspections by the FDA to determine compliance with applicable regulations, and these inspections may include the manufacturing facilities
of some of our subcontractors. Failure by manufacturers or their suppliers to comply with applicable regulatory requirements can result
in enforcement action by the FDA or other regulatory authorities. Potential FDA enforcement actions include:
| |
● |
untitled
letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
● |
unanticipated
expenditures to address or defend such actions |
| |
● |
customer
notifications for repair, replacement, refunds; |
| |
● |
recall,
detention or seizure of our products; |
| |
● |
operating
restrictions or partial suspension or total shutdown of production; |
| |
● |
refusing
or delaying our requests for 510(k) clearance or premarket approval of new products or modified products; |
| |
● |
operating
restrictions; |
| |
● |
withdrawing
510(k) clearances on PMA approvals that have already been granted; |
| |
● |
refusal
to grant export approval for our products; or |
| |
● |
criminal
prosecution. |
In
addition, other government authorities influence the success of our business, including the availability of adequate reimbursement from
third party payors, including government programs such as Medicare and Medicaid. Medicare and Medicaid reimbursement policies can also
influence corresponding policies of private insurers and managed care providers, which can further affect our business.
Combination
Products
A
combination product is the combination of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic,
that are combined or mixed and produced as a single entity; packaged together in a single package or as a unit; or a drug, device, or
biological product packaged separately that according to its investigational plan or proposed labeling, is intended for use only with
an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication,
or effect.
To
determine which FDA center or centers will review a combination product candidate submission, companies may submit a request for assignment
to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on the
FDA’s experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also
may submit a formal “Request for Designation” to the FDA Office of Combination Products. The Office of Combination Products
will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation.
The
FDA will determine which center or centers within the FDA will review the product candidate and under what legal authority the product
candidate will be reviewed. Depending on how the FDA views the product candidates that are developed, the FDA may have aspects of the
product candidate reviewed by CBER, CDRH, or CDER, though one center will be designated as the center with primary jurisdiction, based
on the product candidate’s primary mode of action. The FDA determines the primary mode of action based on the single mode of action
that provides the most important therapeutic action of the combination product candidate. This would be the mode of action expected to
make the greatest contribution to the overall intended therapeutic effects of the combination product candidate. The review of such combination
product candidates is often complex and time consuming, as the FDA may select the combination product candidate to be reviewed and regulated
by one, or multiple FDA centers identified above, which could affect the path to regulatory clearance or approval. Furthermore, the FDA
may also require submission of separate applications to multiple centers.
Once
commercialized, manufacturers of combination products must generally comply with the applicable regulations governing each constituent
part. For example, in January 2013, the FDA finalized 21 CFR Part 4, “Current Good Manufacturing Practice Requirements for Combination
Products”, which was effective July 22, 2013. Associated guidance was also issued in January 2017. Both the rule and guidance reiterate
that combination product manufacturers are responsible for compliance with both biologic and device cGMPs when engaging in manufacturing
both constituent parts. The guidance allows the use of an abbreviated approach as well. Manufacturers of combination products also must
comply with post marketing safety reporting, or PMSR, requirements in accordance with 21 CFR Part 4.
We
have been informed by the FDA that our esophageal implant is a combination biologic/device product. Biological products must satisfy
the requirements of the PHS Act and the FDCA and their implementing regulations. The lead reviewing FDA Center will be the Center for
Biologics Evaluation and Research or CBER. The CBER may choose to consult or collaborate with the FDA’s Center for Devices and
Radiological Health, or CDRH, with respect to the characteristics of the synthetic scaffold component of our product based on the CBER’s
determination of need for such assistance. Because the CBER is the lead, in order for our esophageal implant to be legally marketed in
the U.S., the product must have a BLA approved by the FDA.
We
discuss both the CBER and the CDRH regulatory paradigms below, as potential future products may implicate elements of each, largely at
the CBER’s discretion to involve the CDRH in the review and approval process.
The
BLA Approval Process
The
basic steps for obtaining FDA approval of a BLA to market a biopharmaceutical, or biologic product in the U.S. include:
| |
● |
completion
of preclinical laboratory tests, animal studies and formulation studies under the FDA’s GLP regulations; |
| |
|
|
| |
● |
submission
to the FDA of an IND application, for human clinical testing, which must become effective before human clinical trials may begin
and which must include Institutional Review Board, or IRB, approval at each clinical site before the trials may be initiated; |
| |
|
|
| |
● |
performance
of adequate and well-controlled clinical trials in accordance with Good Clinical Practices, or GLP, to establish the safety, purity,
and potency of the product for each indication; |
| |
|
|
| |
● |
submission
to the FDA of a BLA, which contains detailed information about the chemistry, manufacturing and controls for the product, reports
of the outcomes of the clinical trials, and proposed labeling and packaging for the product; |
| |
|
|
| |
● |
the
FDA’s acceptance of the BLA for filing; |
| |
|
|
| |
● |
satisfactory
review of the contents of the BLA by the FDA, including the satisfactory resolution of any questions raised during the review or
by the advisory committee, if applicable; |
| |
|
|
| |
● |
satisfactory
completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance
with cGMP regulations, to assure that the facilities, methods and controls are adequate to ensure the product’s identity, strength,
quality and purity; and |
| |
|
|
| |
● |
FDA
approval of the BLA. |
In
order to obtain approval to market a biological product in the United States, a marketing application must be submitted to the FDA that
provides sufficient data establishing the safety, purity and potency of the proposed biological product for its intended indication.
The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous
results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls
and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness
of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval,
the data submitted must be sufficient in quality and quantity to establish the safety, purity and potency of the biological product to
the satisfaction of the FDA.
The
results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical
tests conducted on the chemistry of the drug, proposed labeling, and other relevant information are submitted to the FDA as part of a
BLA requesting approval to market the product. The submission of a BLA is subject to the payment of user fees; a waiver of such fees
may be obtained under certain limited circumstances. The FDA initially reviews all BLAs submitted to ensure that they are sufficiently
complete for substantive review before it accepts them for filing. The FDA generally completes this preliminary review within 60 calendar
days. The FDA may request additional information rather than accept a BLA for filing. In this event, the BLA must be resubmitted with
the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission
is accepted for filing, the FDA begins an in-depth substantive review. FDA may refer the BLA to an advisory committee for review, evaluation
and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation
of an advisory committee, but it generally follows such recommendations. The approval process is lengthy and often difficult, and the
FDA may refuse to approve a BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data
and information. Even if such data and information are submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria
for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret
the same data. FDA reviews a BLA to determine, among other things whether the product is safe, pure and potent and the facility in which
it is manufactured, processed, packed or held meets standards designed to assure the product’s continued safety, purity and potency.
Before approving a BLA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA may issue a complete
response letter, which may require additional clinical or other data or impose other conditions that must be met in order to secure final
approval of the BLA, or an approval letter following satisfactory completion of all aspects of the review process.
BLAs
may receive either standard or priority review. Under current FDA review goals, standard review of an original BLA will be 10 months
from the date that the BLA is filed. A biologic representing a significant improvement in treatment, prevention or diagnosis of disease
may receive a priority review of six months. Priority review does not change the standards for approval but may expedite the approval
process.
If
the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will either issue “not
approvable” letter or an “approvable” letter. A “not approvable” letter means that the FDA refuses to approve
the application because the BLA or manufacturing facilities do not satisfy the regulatory criteria for approval. An “approvable”
letter means that the FDA considers the BLA and manufacturing facilities to be favorable, but the letter will outline the deficiencies
and provide the applicant with an opportunity to submit additional information or data to address the deficiencies. If and when those
conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. If a product receives regulatory
approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could
restrict the commercial value of the product. In addition, the FDA may require a sponsor to conduct Phase IV testing which involves clinical
trials designed to further assess a drug’s safety and effectiveness after BLA approval and may require testing and surveillance
programs to monitor the safety of approved products which have been commercialized. Notwithstanding the submission of any requested additional
information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. Separate approval
is required for each proposed indication. If we want to expand the use of an approved product, we will have to design additional clinical
trials, submit the trial designs to the FDA for review and complete those trials successfully.
The
Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted in 2012, made permanent the Pediatric Research Equity
Act, or PREA, which requires a sponsor to conduct pediatric studies for most biologics with a new active ingredient, new indication,
new dosage form, new dosing regimen or new route of administration. Under PREA, BLAs and supplements thereto, must contain a pediatric
assessment unless the sponsor has received a deferral or waiver. The required assessment must assess the safety and effectiveness of
the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric
subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of pediatric studies for some or
all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the biologic is ready for
approval for use in adults before pediatric studies are complete or that additional safety or effectiveness data needs to be collected
before pediatric studies can begin. After April 2013, the FDA must send a non-compliance letter to any sponsor that fails to submit a
required pediatric assessment within specified deadlines or fails to submit a timely request for approval of a pediatric formulation,
if required.
Priority
or Expedited Review Pathways for BLAs
Companies
may seek fast track designation for their products. Fast track products are those that are intended for the treatment of a serious or
life-threatening condition and that demonstrate the potential to address unmet medical needs for such a condition. If awarded, the fast
track designation applies to the product only for the indication for which the designation was received. Fast track products are eligible
for two means of potentially expediting product development and FDA review of BLAs. First, a fast track product may be approved on the
basis of either a clinical endpoint or a surrogate endpoint that is reasonably likely to predict clinical benefit. Approvals of this
kind may be subject to requirements for appropriate post-approval studies to validate the surrogate endpoint or otherwise confirm the
effect on the clinical endpoint, and to certain other conditions. Second, if the FDA determines after review of preliminary clinical
data submitted by the sponsor that a fast track product may be effective, it may begin review of portions of a BLA before the sponsor
submits the complete BLA, thereby accelerating the date on which review of a portion of the BLA can begin. There can be no assurance
that any of our other products will receive designation as fast track products. And even if they are designated as fast track products,
we cannot assure you that our products will be reviewed or approved more expeditiously for their fast track indications than would otherwise
have been the case or will be approved promptly, or at all. Furthermore, the FDA can revoke fast track status at any time.
In
addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful
therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well-controlled
clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical
benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval,
the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-approval clinical
trials to verify and further define the drug’s clinical benefit and safety profile. There can be no assurance that any of our products
will receive accelerated approval. Even if accelerated approval is granted, the FDA may withdraw such approval if the sponsor fails to
conduct the required post-approval clinical trials, or if the post-approval clinical trials fail to confirm the early benefits seen during
the accelerated approval process.
Fast-Track
designation and accelerated approval should be distinguished from priority review although products awarded fast track status may also
be eligible for priority review. Products regulated by the CBER may receive priority review if they provide significant improvement in
the safety or effectiveness of the treatment, diagnosis, or prevention of a serious or life-threatening disease. Products awarded priority
review are given abbreviated review goals by the agency. Under the Prescription Drug User Fee Act of 2007, the agency has agreed to the
performance goal of reviewing products awarded priority review within six months, whereas products under standard review receive a ten-month
target. The review process, however, is often significantly extended by FDA requests for additional information or clarification regarding
information already provided in the submission. Priority review is requested at the time the BLA is submitted, and the FDA makes a decision
as part of the agency’s review of the application for filing. We plan to seek priority review for our trachea transplant products
but cannot guarantee that the FDA will grant the designation and cannot predict if awarded, what impact, if any, it will have on the
review time for approval of our product.
We
intend to request Fast Track status, Breakthrough Therapy designation, Regenerative Medicine Advanced Therapy, or RMAT, designation,
Accelerated Approval and Priority Review. If we are awarded any of these designations, combined with our Orphan Drug designations, discussed
below, we believe that our future clinical trial designs and approval pathway may be streamlined and expedited. Although, if granted,
Fast-Track designation, accelerated approval, and priority review may expedite the approval process, they do not change the standards
for approval. On September 30, 2020, Congress provided a short-term extension of the rare pediatric disease Priority Review Voucher Program.
According to the current statutory sunset provisions:
| |
1) |
After
December 11, 2020, the FDA may only award a voucher for an approved RPD product application if the sponsor has RPD designation for
the drug and that designation was granted by December 11, 2020. |
| |
|
|
| |
2) |
After
December 11, 2022, the FDA may not award any RPD priority review vouchers. |
The
Creating Hope Reauthorization Act, which was received in the Senate on September 30, 2020, proposes to replace those cutoffs with “September
30, 2024” and “September 30, 2026”, respectively, thus extending the authorized period for RPD designation and granting
of RPD priority review vouchers from the 21st Century Cures Act by four years. We cannot be certain that this extension will
be granted.
Clinical
Trials
BLAs
generally require clinical data in order for FDA review and approval. Clinical trials are subject to extensive monitoring, recordkeeping
and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and
must comply with FDA regulations, including but not limited to those relating to GCP. Adverse events must be reported and investigated
timely. To conduct a clinical trial, a company is also required to obtain the patients’ informed consent in form and substance
that complies with both FDA requirements and state and federal privacy and human subject protection regulations. The sponsor, the FDA
or the IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to trial subjects outweigh
the anticipated benefits. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part
of the IND. In addition, an IRB at each site at which the trial is conducted must approve the protocol and any amendments. Foreign studies
performed under an IND must meet the same requirements that apply to U.S. studies. The FDA will accept a foreign clinical trial not conducted
under an IND only if the trial is well-designed, well-conducted, performed by qualified investigators in accordance with international
principles for GCP, and conforms to the ethical principles contained in the Declaration of Helsinki, or with the laws and regulations
of the country in which the research was conducted, whichever provides greater protection of the human subjects. The FDA, however, has
substantial discretion in deciding whether to accept data from foreign non-IND clinical trials.
Clinical
trials involving biopharmaceutical products are typically conducted in three sequential phases. The phases may overlap or be combined.
A fourth, or post-approval, phase may include additional clinical trials. These phases are described generally below. Briefly, the phases
of clinical development generally include the following:
| |
● |
Phase
I. Phase I clinical trials involve the initial introduction of the medicine into human subjects to determine the adverse effects
associated with increasing doses. Such Phase I studies frequently are highly abbreviated or combined with Phase II studies (as outlined
below). |
| |
|
|
| |
● |
Phase
II. Phase II clinical trials usually involve studies in a limited patient population to evaluate the efficacy of the product
for specific, targeted indications to identify possible adverse effects and safety risks. |
| |
|
|
| |
● |
Phase
III. If the biologic is found to be potentially effective and to have an acceptable safety profile in Phase II (or sometimes
Phase I) trials, the clinical trial program will be expanded to further demonstrate clinical efficacy, optimal dosage and safety
within an expanded patient population at geographically dispersed clinical trial sites. As noted, the exact number of subjects needed,
the duration of clinical follow-up, and the endpoints by which safety and efficacy are demonstrated are based on the condition being
treated. |
| |
|
|
| |
● |
Post-Approval
(Phase IV). Post-approval clinical trials are required of or agreed to by a sponsor as a condition of, or subsequent to marketing
approval. Further, if the FDA becomes aware of new safety information about an approved product, it is authorized to require post
approval trials of the biological product. These trials are used to gain additional experience from the treatment of patients in
the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval
regulations. If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company
may be able to use the data from these clinical trials to meet all or part of any Phase IV clinical trial requirement. These clinical
trials are often referred to as Phase III/IV post approval clinical trials. Failure to promptly conduct Phase IV clinical trials
could result in withdrawal of approval for products approved under accelerated approval regulations. |
Medical
devices, however, typically rely on one or a few pivotal studies rather than Phase I, II, and III clinical trials.
During
the development of a new medical product, sponsors are given opportunities to meet with the FDA at certain points. These points may be
prior to submission of an IND or IDE, at the end of Phase II, and before a BLA or PMA is submitted. Meetings at other times may be requested.
These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide
advice, and for the sponsor and FDA to reach agreement on the next phase of development. Sponsors typically use the end of Phase II meeting
to discuss their Phase II clinical results and present their plans for the pivotal Phase III clinical trial that they believe will support
approval of the new biologic. Similarly, sponsors typically use the end of feasibility studies to do the same for planning for their
pivotal trial or trials for a medical device.
Concurrent
with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry
and physical characteristics of a biologic and finalize a process for manufacturing the product in commercial quantities in accordance
with cGMP requirements. For biologics, the manufacturing process must be capable of consistently producing quality batches of the product
candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the
final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate
that the product candidate does not undergo unacceptable deterioration over its shelf life. Before approving a BLA, the FDA typically
will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines
that the manufacturing processes and facilities are in full compliance with cGMP requirements and adequate to assure consistent production
of the product within required specifications. The PHSA in particular emphasizes the importance of manufacturing control for products
like biologics whose attributes cannot be precisely defined.
Based
on the FDA’s approval of our clinical trial for any condition that requires removal of part of the esophagus, we believe that we
are able to pursue the treatment of multiple diseases, injuries or birth defects with a single clinical trial. As a result, we believe
that this clinical trial will advance our esophageal implant for numerous indications including to treat esophageal cancer, Barrett esophagus,
fistulas, traumatic injury to the esophagus and birth defects in the esophagus. Compared to developing treatments for a single underlying
medical condition, we believe that addressing multiple medical conditions in a single clinical trial has the potential to significantly
reduce our costs to expand the market for our products. Based on discussions with the FDA, we also expect clinical trials for our esophageal
implant product candidates to be conducted in two sequential phases:
| |
● |
An
initial trial that combines both phase 1 and phase 2 into a single trial. This trial has already been approved by the FDA. |
| |
|
|
| |
● |
If
successful, the initial trial would be followed by a phase 2 Registration, or Pivotal Trial, to test the product candidate’s
safety and efficacy in a larger patient population. We believe that the nature of the our esophageal implant and the sizes of their
targeted patient populations would lead to a small number of patients in this trial, relative to most biotechnology clinical trials. |
As
with any clinical trial, clinical testing of our esophageal implant may not be completed successfully within any specified time period,
if at all. The FDA closely monitors the progress of each phase of clinical trials that are conducted under an IND and may, at its discretion,
reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of
the risk/benefit ratio to the patient. The FDA or the sponsor may suspend or terminate clinical trials at any time for various reasons,
including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional
pre-clinical studies or clinical trials be conducted as a condition to product approval.
We
will submit a BLA once we have sufficient data from the clinical trials to assess the safety and efficacy of our esophageal implant.
We estimate that this process may span a period of three to six years, or longer, considering the uncertainty of a successful clinical
trial. We anticipate approvals in countries outside of the United States may be shorter, however, we can give no assurance of such approvals.
Post-Approval
Requirements
After
BLA approval is obtained, companies are required to comply with a number of post-approval requirements relating to manufacturing, labeling,
packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. For example, as a condition of approval
of a BLA, the FDA may require post-approval testing and surveillance to monitor the product’s safety or efficacy. In addition,
holders of an approved BLA are required to keep extensive records, to report certain adverse reactions and production deviations and
problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional
labeling for their products. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory
authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered,
we could be subject to administrative or judicially imposed sanctions or other setbacks. Accordingly, manufacturers must continue to
expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory
compliance.
Specifically,
our products could be subject to voluntary recall if we or the FDA determine, for any reason, that our products pose a risk of injury
or are otherwise defective. Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our device would
cause serious adverse health consequences or death. In addition, the FDA could suspend the marketing of or withdraw a previously approved
product from the market upon receipt of newly discovered information regarding the drug’s safety or effectiveness.
Orphan
Drug Designation
In
November 2016, we were granted Orphan Drug Designation for our esophageal implant by the FDA to restore the structure and function of
the esophagus subsequent to esophageal damage due to cancer, injury or congenital abnormalities. We also were granted Orphan Drug Designation
for trachea on September 4, 2014.
The
Orphan Drug Act provides incentives to manufacturers to develop and market drugs and biologics for rare diseases and conditions affecting
fewer than 200,000 persons in the U.S. at the time of application for orphan drug designation, or more than 200,000 individuals in the
U.S. and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in
the U.S. for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested
before submitting a new drug application, or NDA, or BLA. After the FDA grants orphan product designation, the identity of the therapeutic
agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or
shorten the duration of the regulatory review and approval process. The first developer to receive FDA marketing approval for an orphan
biologic is entitled to a seven year exclusive marketing period in the U.S. for that product as well as a waiver of the BLA user fee.
The exclusivity prevents FDA approval of another application for the same product for the same indication for a period of seven years,
except in limited circumstances where there is a change in formulation in the original product and the second product has been proven
to be clinically superior to the first. In addition, Orphan Drug Designation provides a seven-year marketing exclusivity period against
competition in the U.S. from the date of a product’s approval for marketing. This exclusivity would be in addition to any exclusivity
we may obtain from our patents. Additionally, orphan designation provides certain incentives, including tax credits and a waiver of the
Biologics License Application, or BLA, fee. We also plan to apply for Orphan Drug Designation for our esophageal implant in Europe. Orphan
Drug Designation in Europe would provide market exclusivity in Europe for a period of ten years from the date of the product’s
approval for marketing.
International
We
plan to seek required regulatory approvals and comply with extensive regulations governing product safety, quality, manufacturing and
reimbursement processes in order to market our products in other major foreign markets.
In
addition to having large patient populations, both the E.U. and some countries in Asia allow for “conditional approval” for
product candidates like ours. Conditional approval is country specific but, in general, it would allow us to market our products, and
obtain revenue from the sales of them, after successful phase 2 results. Conditional approval is granted subject to the regulatory authority
being able to rescind the approval if something goes wrong in as more patients get treated. Hence, it is possible that we could see revenue
in either Asia or the E.U. before we see revenue in the U.S.
The
regulation of our products in the Asian and European markets, and in other foreign markets varies significantly from one jurisdiction
to another. The classification of the particular products and related approval or CE marking procedures can involve additional product
testing and additional administrative review periods. The time required to obtain these foreign approvals or to CE mark our products
may be longer or shorter than that required in the U.S., and requirements for approval may differ from the FDA requirements. Regulatory
approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one
country may negatively impact the regulatory process in others.
Legislation
similar to the Orphan Drug Act has been enacted in other jurisdictions, including the E.U. The orphan legislation in the E.U. is available
for therapies addressing conditions that affect five or fewer out of 10,000 persons. The marketing exclusivity period is for ten years,
although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product is
sufficiently profitable not to justify maintenance of market exclusivity. We intend to apply for orphan drug-designation for our esophageal
implant in Europe.
We
have also formed a subsidiary in Hong Kong, Harvard Apparatus Regenerative Technology Limited, as we continue to assess the market and
regulatory approval pathway in China as to our product candidates. We have other subsidiaries in the U.K. and Germany. Any development
and capital raising efforts in China may include a joint venture in relation to our Hong Kong subsidiary, and would also involve a number
of commercial variables, including rights and obligations pertaining to licensing, development and financing, among others. Our failure
to receive or obtain such clearances or approvals on a timely basis or at all, whether that be in the U.S., China or otherwise, would
have an adverse effect on our results of operations.
Employees
and Human Capital Resources
As
of December 31, 2023, our consolidated business employed 18 individuals. Our employees are located in the U.S. and Asia and the laws
regarding employee relationships are different by jurisdiction. None of our employees are unionized. In general, we consider our
relations with our employees to be good. Our employees are highly skilled, and many hold advanced degrees. Our future performance
depends significantly upon the continued service of our key scientific, technical and senior management personnel and our continued
ability to attract and retain highly skilled employees. We have taken proactive steps throughout the COVID-19 pandemic to protect
the health and safety of our employees. We expect to continue to implement these measures until we determine that the COVID-19
pandemic is adequately contained for purposes of our business. We may take further actions, in compliance with all appropriate
government regulations, that we determine to be in the best interest of our employees.
Competition
We
are not aware of any companies whose products are directly competitive with our cell-seeded biocompatible synthetic-scaffold system.
However, in our key markets we may in the future compete with multiple pharmaceutical, biotechnology, and medical device companies, many
of which have substantially greater financial, technological, research and development, marketing and personnel resources than we do.
In addition, there are many academic and clinical centers that are developing regenerative technologies that may one day become competitors
of ours.
We
expect that other products will compete with our products and potential products based on efficacy, safety, cost, and intellectual property
positions. While we believe that these will be the primary competitive factors, other factors include, in certain instances, obtaining
marketing exclusivity under the Orphan Drug Act, availability of supply, manufacturing, marketing and sales expertise and capability,
and reimbursement coverage.
Information
about our Executive Officers
The
following table shows information about our executive officers as of March 18, 2024:
| Name |
|
Age |
|
Position(s) |
| Junli
(Jerry) He |
|
49 |
|
Chief
Executive Officer |
| Hong
Yu |
|
51 |
|
President |
| Dr.
William Fodor |
|
65 |
|
Chief
Scientific Officer |
| Joseph
Damasio, Jr. |
|
49 |
|
Chief
Financial Officer |
Junli
(Jerry) He – Chairman and Chief Executive Officer
Mr. He was appointed as our Chairman and Chief Executive Officer (CEO)
on March 1, 2023. He has served as a member of our Board of Directors since September 1, 2021. Mr. He serves as the Executive Vice Chairman
of Bright Scholar Holdings and has been in that position since January 2019. Prior to the promotion, Mr. He had served as the CEO of Bright
Scholar. Prior to joining Bright Scholar, Mr. He was a Managing Director at Tstone Corp, and served as Chief Financial Officer, CEO and
a director of Noah Education Holdings Ltd., a former NYSE listed private education services provider in China. Mr. He was a portfolio
manager at Morgan Stanley Global Wealth Management from June 2008 to June 2009 and was employed by Bear Stearns from November 2006 to
May 2008. Mr. He obtained a bachelor’s degree in science from Peking University and an M.B.A. with Honors from the University of
Chicago, Booth School of Business. Mr. He is also a Certified Financial Analyst (CFA) charter holder.
Hong
Yu, BS, MS, CFA – President
Mr.
Yu has served as our President since May 31, 2018 and has raised over $20 million in capital for Harvard Apparatus Regenerative Technology.
Mr. Yu is a seasoned executive with extensive experience in fundraising, strategic analytics, wealth management, and investment research.
Prior to Harvard Apparatus Regenerative Technology, Mr. Yu was a Senior Vice President at Bank of America, where he was employed for nearly
20 years. During his career, Mr. Yu has developed an expertise in matching emerging companies with cross-border investors. Mr. Yu holds
a B.S. degree from Peking University (Beijing, China), and a M.S. degree from University of Illinois (Chicago, IL). Mr. Yu is a Chartered
Financial Analyst.
Dr.
William Fodor – Chief Scientific Officer
Dr.
William Fodor has served as our Chief Scientific Officer since July 2017. On July 2, 2018, Dr. Fodor became an employee of Harvard Apparatus
Regenerative Technology after serving as a consultant to the Company. Dr. Fodor was a founding scientist at Alexion Pharmaceuticals,
where he served as an executive management team member and Senior Director of the Cell/Tissue Engineering, Transgenic Animal and Transplant
Programs. He has also served as an Associate Professor at the University of Connecticut Department of Molecular Cell Biology and the
Center for Regenerative Biology, extending research areas into stem cells and cell engineering. Dr. Fodor was Senior Director of Product
Development at ViaCell Inc., leading programs in hematopoietic stem cell process development and manufacturing, mesenchymal stem cell
basic research and manufacturing for cardiac repair and pancreatic stem cell research. He was a consultant for the biotechnology industry,
serving clients in stem cell research, gene therapy, stem cell manufacturing and stem cell genome engineering. Dr. Fodor has expertise
in programs targeting transplant immunology, hematopoiesis, cardiac repair, stem cell potency, gene therapy for liver diseases, tissue
engineering, design and oversight of pre-clinical non-Good Laboratory Practices (GLP) and GLP animal models and IND Applications
(Pre-clinical and Chemistry, Manufacturing and Control (CMC) Modules). Dr. Fodor earned a PhD. In genetics from Ohio State University.
He completed post-doctoral work at Yale University School of Medicine in the department of immunobiology, investigating the regulation
of Major histocompatibility complex (MHC) class I and MHC class II genes in the histocompatibility complex.
Joseph
Damasio, Jr. – Chief Financial Officer
Mr.
Damasio has served as our Chief Financial Officer since August 8, 2022. He has over 20 years of finance and accounting experience. Prior
to joining our company, he was Vice President of Finance at Inhibikase Therapeutics, a publicly-traded clinical stage biopharmaceutical
company, since October 2021. Before joining Inhibikase, Mr. Damasio was Controller at Cue Biopharma from June 2020 to October 2021, Controller
at XL Fleet from February 2019 to June 2020, and Chief Financial Officer at Pressure BioSciences, Inc. from April 2017 to February 2019.
Mr. Damasio earned a bachelor’s degree in accounting, with honors, from the University of Massachusetts. He holds an MBA and MSF
from Boston College and is a Certified Public Accountant in Massachusetts.
Available
Information and Website
Our
website address is www.hregen.com. Our Annual Reports on Form 10-K, our Quarterly Reports on Form 10-Q, Current Reports on Form
8-K, and exhibits and amendments to those reports filed or furnished with the Securities and Exchange Commission, or SEC, pursuant to
Section 13(a) of the Exchange Act are available for review on our website and the SEC website at www.sec.gov. Any such materials that
we file with, or furnish to, the SEC in the future will be available on our website as soon as reasonably practicable after they are
electronically filed with, or furnished to, the SEC. The information on our website is not incorporated by reference into this Annual
Report on Form 10-K.
Item
1A. Risk Factors.
Summary
of Risk Factors
Below
is a summary of the principal factors that make an investment in our common stock speculative or risky. This summary does not address
all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face,
can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in
this Annual Report on Form 10-K and our other filings with the SEC before making an investment decision regarding our common stock.
| ● |
Our
audited financial statements for the year ended December 31, 2023 contain a going concern qualification. Our financial status creates
doubt whether we will continue as a going concern. We will need additional funds in the near future and our operations will be adversely
affected if we are unable to obtain needed funding. |
| ● |
We
have generated an insignificant amount of revenue from commercial operations to date and have an accumulated deficit. We anticipate
that we will incur losses for the foreseeable future. We may never achieve or sustain profitability. |
| ● |
The
COVID-19 pandemic could continue to adversely impact our business, including clinical trials. |
| ● |
Our
product candidates are in an early stage of development. If we are unable to develop or market any of our product candidates, our
financial condition will be negatively affected, and we may have to curtail or cease our operations. |
| ● |
Our
product candidates will subject us to liability exposure. |
| ● |
The
results of our clinical trials or pre-clinical development efforts may not support our product candidates claims or may result in
the discovery of adverse side effects. |
| ● |
If
we fail to obtain, or experience significant delays in obtaining, regulatory approvals in the U.S., China or the E.U. for our product
candidates, including those for the esophagus and airways, or are unable to maintain such clearances or approvals for our product
candidates, our ability to commercially distribute and market these products would be adversely impacted. |
| ● |
Even
if our product candidates are cleared or approved by regulatory authorities, if we or our suppliers fail to comply with ongoing FDA
or other foreign regulatory authority requirements, or if we experience unanticipated problems with our product candidates, these
product candidates could be subject to restrictions or withdrawal from the market. |
| ● |
General
market conditions, including the effects of Russia’s invasion of Ukraine and attendant economic sanctions, high inflation,
and rising interest rates as well as the effects of laws and regulations on foreign investment in the United States under the jurisdiction
of the Committee on Foreign Investment in the United States (CFIUS), and other agencies and related regulations, including the Foreign
Investment Risk Review Modernization Act (FIRRMA), adopted in August 2018, may make it difficult for us to seek financing from the
capital markets. |
| ● |
Our
principal stockholders hold a significant percentage of our voting power and will be able to exert significant control over us. |
| ● |
We
do not intend to pay cash dividends on our common stock. |
Risk
Factors
The
following factors should be reviewed carefully, in conjunction with the other information contained in this Annual Report on Form 10-K.
As previously discussed, our actual results could differ materially from our forward-looking statements. Our business faces a variety
of risks. We describe below what we believe are currently the material risks and uncertainties we face, but they are not the only risks
and uncertainties we face. Additional risks and uncertainties of which we are unaware, or that we currently believe are not material,
may also become important factors that adversely affect our business. In addition, past financial performance may not be a reliable indicator
of future performance and historical trends should not be used to anticipate results or trends in future periods. If any of the following
risks and uncertainties develops into actual events, these events could have a material adverse effect on our business, financial condition
or results of operations. In such case, the trading price of our common stock could decline, and you may lose all or part of your investment
in our securities. The risk factors generally have been separated into three groups: (i) risks relating to our business, (ii) risks relating
to the Separation and (iii) risks relating to our common stock. These risk factors should be read in conjunction with the other information
in this Annual Report on Form 10-K.
Risks
Relating to Our Financial Position, Need for Capital and Operating Risks
Our
audited financial statements for the year ended December 31, 2023 contain a going concern qualification. Our financial status creates
doubt whether we will continue as a going concern. We will need additional funds in the near future and our operations will be adversely
affected if we are unable to obtain needed funding.
We
ended the year 2023 with approximately $0.4 million of operating cash on-hand and received debt financing of $0.5 million in
gross proceeds subsequent to December 31, 2023 and will need to raise additional capital in the first quarter and beyond to
fund operations. If we do not raise additional capital from outside sources during the first quarter of 2024, we may be
forced to further curtail or cease our operations. Based on these circumstances, our ability to continue as a going concern is at
risk and our independent registered public accounting firm included a “going concern” explanatory paragraph as to our
ability to continue as a going concern in their audit report dated March 28, 2024, included in this Form 10-K. Our cash requirements
and cash resources will vary significantly depending upon the timing, and the financial and other resources that will be required to
complete ongoing development and pre-clinical and clinical testing of our product candidates, regulatory efforts and collaborative
arrangements necessary for our product candidates that are currently under development. In addition to development and other costs,
we expect to incur capital expenditures from time to time. These capital expenditures will be influenced by our regulatory
compliance efforts, our success, if any, at developing collaborative arrangements with strategic partners, our needs for additional
facilities and capital equipment and the growth, if any, of our business in general. We will require additional funding to continue
our anticipated operations and support our capital and operating needs. We are currently seeking and will continue to seek
financings from other existing and/or new investors to raise necessary funds through a combination of public or private equity
offerings. We may also pursue debt financings, other financing mechanisms, strategic collaborations and licensing arrangements. We
may not be able to obtain additional financing on terms favorable to us, if at all. In addition, general market conditions,
including the effects of Russia’s invasion of Ukraine and attendant economic sanctions, high inflation, rising interest rates
and the COVID-19 pandemic on financial markets, as well as the effects of laws and regulations on foreign investment in the United
States under the jurisdiction of the Committee on Foreign Investment in the United States (CFIUS), and other agencies and related
regulations, including the Foreign Investment Risk Review Modernization Act (FIRRMA), adopted in August 2018, may make it difficult
for us to seek financing from the capital markets.
Any
additional equity financings could result in significant dilution to our stockholders and possible restrictions on subsequent financings.
Debt financing, if available, could result in agreements that include covenants limiting or restricting our ability to take certain actions,
such as incurring additional debt, making capital expenditures or paying dividends. Other financing mechanisms may involve selling intellectual
property rights, payment of royalties or participation in our revenue or cash flow. In addition, in order to raise additional funds through
strategic collaborations or licensing arrangements, we may be required to relinquish certain rights to some or all of our technologies
or product candidates. If we cannot raise funds or engage strategic partners on acceptable terms when needed, we may not be able to continue
our research and development activities, develop or enhance our product candidates, take advantage of future opportunities, grow our
business, respond to competitive pressures or unanticipated requirements, or at worst may be forced to curtail or cease our operations.
We
have generated insignificant revenue to date and have an accumulated deficit. We anticipate that we will incur losses for the foreseeable
future. We may never achieve or sustain profitability.
We
have generated insignificant revenues to date, and we have generated no revenues from sales of any clinical product candidates, and,
as of December 31, 2023, we had an accumulated deficit of approximately $92.0 million. We expect to continue to experience losses in
the foreseeable future due to our limited anticipated revenues and significant anticipated expenses. We do not anticipate that we will
achieve meaningful revenues for the foreseeable future. In addition, we expect that we will continue to incur significant operating expenses
as we continue to focus on additional research and development, preclinical testing, clinical testing and regulatory review and/or approvals
of our product candidates and technologies. As a result, we cannot predict when, if ever, we might achieve profitability and cannot be
certain that we will be able to sustain profitability, if achieved.
Our
product candidates are in an early stage of development. If we are unable to develop or market any of our product candidates, our financial
condition will be negatively affected, and we may have to curtail or cease our operations.
We
are in the early stage of product development. Investors must evaluate us in light of the uncertainties and complexities affecting an
early-stage biotechnology company. Our product candidates require additional research and development, preclinical testing, clinical
testing and regulatory review and/or approvals or clearances before marketing. In addition, we may not succeed in developing new products
as an alternative to our existing portfolio of product candidates. If we fail to successfully develop and commercialize our product candidates,
including our esophageal or airway product candidates, our financial condition may be negatively affected, and we may have to curtail
or cease our operations.
We
have a limited operating history and it is difficult to predict our future growth and operating results.
We
have a limited operating history and limited operations and assets. Accordingly, investors should consider our prospects in light of
the costs, uncertainties, delays and difficulties encountered by companies in the early stage of development, particularly companies
in new and evolving markets, such as bioengineered organ implants, and regenerative medicine. These risks include, but are not limited
to, unforeseen capital requirements, delays in obtaining regulatory approvals, failure to gain market acceptance and competition from
foreseen and unforeseen sources. As such, our development timelines have been and may continue to be subject to delay that could negatively
affect our cash flow and our ability to develop or bring product candidates to market, if at all. Our estimates of patient population
are based on published data and analysis of external databases by third parties and are subject to uncertainty and possible future revision
as they often require inference or extrapolations from one country to another or one patient condition to another. The effect of any
or all of the foregoing could cause a material adverse effect on our business, financial condition or results of operations.
If
we fail to retain key personnel and/or attract satisfactory replacements, we may not be able to compete effectively, which would have
an adverse effect on our operations.
Our
success is highly dependent on the continued services of key management, technical and scientific personnel and collaborators. Our management
and other employees may voluntarily terminate their employment at any time upon short notice. The loss of the services of any member
of our senior management team, including our Chief Executive Officer Jerry He, our President Hong Yu, our Chief Scientific Officer Dr.
William Fodor, and our Chief Financial Officer Joseph Damasio, and our other key scientific, technical and management personnel, may
significantly delay or prevent the achievement of product development and other business objectives. We can give no assurance that we
could find satisfactory replacements for our current and future key scientific and management employees, including recently terminated
executives, on terms that would not be unduly expensive or burdensome to us.
If
our collaborators do not devote sufficient time and resources to successfully carry out their duties or meet expected deadlines, we may
not be able to advance our product candidates in a timely manner or at all.
We
are currently collaborating with multiple academic researchers and clinicians at a variety of research and clinical institutions. Our
success depends in part on the performance of our collaborators. Some collaborators may not be successful in their research and clinical
trials or may not perform their obligations in a timely fashion or in a manner satisfactory to us. Typically, we have limited ability
to control the amount of resources or time our collaborators may devote to our programs or potential product candidates that may be developed
in collaboration with us. Our collaborators frequently depend on outside sources of funding to conduct or complete research and development,
such as grants or other awards. In addition, our academic collaborators may depend on graduate students, medical students, or research
assistants to conduct certain work, and such individuals may not be fully trained or experienced in certain areas, or they may elect
to discontinue their participation in a particular research program, creating an inability to complete ongoing research in a timely and
efficient manner. As a result of these uncertainties, we are unable to control the precise timing and execution of any experiments that
may be conducted.
Although
we have co-development collaboration arrangements with Mayo Clinic and Connecticut Children’s Medical Center, we do not have formal
agreements in place with other collaborators, and most of our collaborators retain the ability to pursue other research, product development
or commercial opportunities that may be directly competitive with our programs. If any of our collaborators elect to prioritize or pursue
other programs in lieu of ours, we may not be able to advance product development programs in an efficient or effective manner, if at
all. If a collaborator is pursuing a competitive program and encounters unexpected financial or capability limitations, they may be motivated
to reduce the priority placed on our programs or delay certain activities related to our programs. Any of these developments could harm
or slow our product and technology development efforts.
Public
perception of ethical and social issues surrounding the use of cell technology may limit or discourage the use of our technologies, which
may reduce the demand for our products and technologies and reduce our revenues.
Our
success will depend in part upon our and our collaborators’ ability to develop therapeutic approaches incorporating, or discovered
through, the use of cells. If bioengineered organ implant technology is perceived negatively by the public for social, ethical, medical
or other reasons, governmental authorities in the U.S. and other countries may call for prohibition of, or limits on, cell-based technologies
and other approaches to bioengineering and tissue engineering. Although our product candidates have not, to date, used the more controversial
stem cells derived from human embryos or fetuses in the human transplant surgeries using our product candidates, claims that human-derived
stem cell technologies are ineffective or unethical may influence public attitudes. The subject of cell and stem cell technologies in
general has at times received negative publicity and aroused public debate in the U.S. and some other countries. Ethical and other concerns
about such cells could materially harm the market acceptance of our product candidates.
Our
products will subject us to liability exposure.
We
face an inherent risk of product liability claims, especially with respect to our products that will be used within the human body, including
the scaffolds we manufacture. Product liability coverage is expensive and sometimes difficult to obtain, if it can be obtained at all.
We may not be able to obtain or maintain insurance at a reasonable cost. We have and in the future may be subject to claims for liabilities
for unsuccessful outcomes of surgeries involving our products, which may include claims relating to patient suffering and death. We may
also be subject to claims for liabilities relating to patients that suffer serious complications or death during or following implantations
involving our products, including the patients who had surgeries utilizing our first-generation scaffold device or our bioreactor technology
or our esophageal implant, or patients that may have surgeries utilizing any of our products in the future. On April 27, 2022, we and
Harvard Bioscience executed a settlement, relating to an ongoing wrongful death lawsuit, which resolved all claims relating to the litigation.
The settlement resulted in the dismissal with prejudice of the wrongful death claim, and neither we nor Harvard Bioscience admitted any
fault or liability in connection with the claim. The settlement also resolved any and all claims by and between the parties and our products
liability insurance carriers, which resulted in the dismissal with prejudice of all claims asserted by or against those carriers, us
and Harvard Bioscience. Our current product liability coverage is $10 million per occurrence and in the aggregate. We will need to increase
our insurance coverage if and when we begin commercializing any of our products. There can be no assurance that existing insurance coverage
will extend to other products in the future. Any product liability insurance coverage may not be sufficient to satisfy all liabilities
resulting from product liability claims. Furthermore, insurance carriers may deny that coverage exists after a claim is made. A successful
claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable items, if at all. If
claims against us substantially exceed our coverage, then our business could be adversely impacted. Regardless of whether we are ultimately
successful in any product liability litigation, such litigation could consume substantial amounts of our financial and managerial resources
and could result in, among others:
| |
● |
significant
awards or judgments against us; |
| |
● |
substantial
litigation costs; |
| |
● |
injury
to our reputation and the reputation of our products; |
| |
● |
withdrawal
of clinical trial participants; and |
| |
● |
adverse
regulatory action. |
Any
of these results would substantially harm our business.
If
restrictions on reimbursements or other conditions imposed by payers limit our customers’ actual or potential financial returns
on our products, our customers may not purchase our products or may reduce their purchases.
Our
customers’ willingness to use our products will depend in part on the extent to which coverage for these products is available
from government payers, private health insurers and other third-party payers. These payers are increasingly challenging the price of
medical products and services. Significant uncertainty exists as to the reimbursement status of newly approved treatments and products
in the fields of biotechnology and regenerative medicine, and coverage and adequate payments may not be available for these treatments
and products. In addition, third-party payers may require additional clinical trial data to establish or continue reimbursement coverage.
These clinical trials, if required, could take years to complete and could be expensive. There can be no assurance that the payers will
agree to continue reimbursement or provide additional coverage based upon these clinical trials. Failure to obtain adequate reimbursement
would result in reduced sales of our products, which could have a material adverse effect on our business, financial condition and results
of operations.
We
depend upon single-source suppliers for the hardware used for our proprietary automatic cell seeder, bioreactor control and acquisition
system. The loss of a single source supplier, or future single-source suppliers we may rely on, or their failure to provide us with an
adequate supply of their products or services on a timely basis, could adversely affect our business.
We
currently have single-source suppliers for certain components that we use for our proprietary automatic cell seeder, bioreactor control
and acquisition systems as well as materials used in scaffolds. We may also rely on other single-source suppliers for critical components
of our products in the future. If we were unable to acquire hardware or other products or services from applicable single-source suppliers,
we could experience a delay in developing and manufacturing our products, which could have a material adverse effect on our business,
financial condition and results of operations.
We
use and generate hazardous materials in our business and must comply with environmental laws and regulations, which can be expensive.
Our
research, development and manufacturing involve the controlled use of hazardous chemicals, and we may incur significant costs as a result
of the need to comply with numerous laws and regulations. For example, certain volatile organic laboratory chemicals we use, such as
fluorinated hydrocarbons, must be disposed of as hazardous waste. We are subject to laws and regulations enforced by the FDA, foreign
health authorities and other regulatory requirements, including the Occupational Safety and Health Act, the Environmental Protection
Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other current and potential federal, state, local
and foreign laws and regulations governing the use, manufacturing, storage, handling and disposal of our products, materials used to
develop and manufacture our products, and resulting waste products. Although we believe that our safety procedures for handling and disposing
of such materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury
from these materials cannot be completely eliminated. In the event of such an accident, our operations could be interrupted. Further,
we could be held liable for any damages that result and any such liability could exceed our resources.
Our
products are novel and will require market acceptance.
Even
if we receive regulatory approvals for the commercial use of our product candidates, their commercial success will depend upon acceptance
by physicians, patients, third party payers such as health insurance companies and other members of the medical community. Market acceptance
of our products is also dependent upon our ability to provide acceptable evidence and the perception of the positive characteristics
of our products relative to existing or future treatment methods, including their safety, efficacy and/or other positive advantages.
If our products fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business. Market acceptance
of, and demand for, any product that we may develop and commercialize will depend on many factors, both within and outside of our control.
If our products receive only limited market acceptance, our business, financial condition and results of operations would be materially
and adversely affected.
Our
long-term growth depends on our ability to develop products for other organs.
Our
growth strategy includes expanding the use of our products in treatments pertaining to organs other than the esophagus and airways, such
as the lungs, gastrointestinal tract, and others. These other organs are more complex than the esophagus and airways. There is no assurance
that we will be able to successfully apply our technologies to these other more complex organs, which might limit our expected growth.
Our
success will depend partly on our ability to operate without infringing on, or misappropriating, the intellectual property or confidentiality
rights of others.
We
may be sued for infringing on the intellectual property or confidentiality rights of others, including the patent rights, trademarks
and trade names and confidential information of third parties. To the extent that any of such claims are valid, if we had utilized, or
were to utilize, such patent applications or patents without an agreement from the owner thereof, it could result in infringement of
the intellectual property rights of the respective owner. Intellectual property and related litigation is costly and the outcome is uncertain.
If we do not prevail in any such intellectual property or related litigation, in addition to any damages we might have to pay, we could
be required to stop the infringing activity, or obtain a license to or design around the intellectual property or confidential information
in question. If we are unable to obtain a required license on acceptable terms or are unable to design around any third-party patent,
we may be unable to sell some of our products and services, which could result in reduced revenue.
We
may be involved in lawsuits to protect or enforce our patents that would be expensive and time consuming.
In
order to protect or enforce our patent and trademark rights, we may initiate litigation against third parties. We may also become subject
to interference proceedings conducted in the patent and trademark offices of various countries to determine the priority of inventions.
The defense and prosecution, if necessary, of intellectual property suits, interference proceedings and related legal and administrative
proceedings would be costly and may divert our technical and management personnel from their normal responsibilities. We may not prevail
in any of these suits should they occur. An adverse determination of any litigation or defense proceedings could put our patents at risk
of being invalidated or interpreted narrowly and could put our patent applications at risk of being rejected and patents not being issued.
Furthermore,
because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some
of our confidential information could be compromised by disclosure during this type of litigation. For example, during the course of
this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments
in the litigation. Securities analysts or investors may perceive these announcements to be negative, which could cause the market price
of our stock to decline.
If
we are unable to effectively protect our intellectual property, third parties may use our technology, which would impair our ability
to compete in our markets.
Our
continued success will depend significantly on our ability to obtain and maintain meaningful patent protection for certain of our products
throughout the world. Patent law relating to the scope of claims in the biotechnology, regenerative medicine, and medical device fields
in which we operate is still evolving. The degree of future protection for our proprietary rights is uncertain. We may rely on patents
to protect a significant part of our intellectual property and to enhance our competitive position. However, our presently pending or
future patent applications may not be accepted and patents might not be issued, and any patent previously issued to us may be challenged,
invalidated, held unenforceable or circumvented. Furthermore, the claims in patents which have been issued or which may be issued to
us in the future may not be sufficiently broad to prevent third parties from producing competing products similar to our products. We
may also operate in countries where we do not have patent rights and in those countries we would not have patent protection. We also
rely on trademarks and trade names in our business. The laws of various foreign countries in which we compete may not protect our intellectual
property to the same extent as do the laws of the U.S. If we fail to obtain adequate patent protection for our proprietary technology,
our ability to be commercially competitive could be materially impaired. It is also possible that our intellectual property may be stolen
via cyber-attacks or similar methods.
In
addition to patent protection, we also rely on protection of trade secrets, know-how and confidential and proprietary information. To
maintain the confidentiality of trade-secrets and proprietary information, we generally seek to enter into confidentiality agreements
with our employees, consultants and strategic partners upon the commencement of a relationship. However, we may not be able to obtain
these agreements in all circumstances in part due to local regulations. In the event of unauthorized use or disclosure of this information,
these agreements, even if obtained, may not provide meaningful protection for our trade-secrets or other confidential information. In
addition, adequate remedies may not exist in the event of unauthorized use or disclosure of this information. The loss or exposure of
our trade secrets and other proprietary information would impair our competitive advantages and could have a materially adverse effect
on our operating results, financial condition and future growth prospects.
Intellectual
property rights do not necessarily address all potential threats to our competitive advantage.
The
degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations,
and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| ● |
we
or any collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications
that we own; |
| ● |
others
may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual
property rights; |
| ● |
it
is possible that our pending patent applications will not lead to issued patents; |
| ● |
our
competitors might conduct research and development activities in the United States and other countries that provide a safe harbor
from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent
rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial
markets; and |
| ● |
we
may not develop additional proprietary technologies that are patentable. |
Our
competitors and potential competitors may have greater resources than we have and may develop products and technologies that are more
effective or commercially attractive than our products and technologies or may develop competing relationships with our key collaborators.
We
expect to compete with multiple pharmaceutical, biotechnology, medical device and scientific research product companies. In addition,
there are many academic and clinical centers that are developing bioengineered or regenerative organ technologies that may one day become
competitors for us. Many of our competitors and potential competitors have substantially greater financial, technological, research and
development, marketing, and personnel resources than we do. We cannot, with any accuracy, forecast when or if these companies are likely
to bring bioengineered organ or regenerative medicine products to market for indications that we are also pursuing. Many of these potential
competitors may be further along in the process of product development and also operate large, company-funded research and development
programs.
We
expect that other products will compete with our current and future products based on efficacy, safety, cost, and intellectual property
positions. While we believe that these will be the primary competitive factors, other factors include obtaining marketing exclusivity
under certain regulations, availability of supply, manufacturing, marketing and sales expertise and capability, and reimbursement coverage.
Our competitors may develop or market products that are more effective or commercially attractive than our current or future products
and may also develop competing relationships with our key collaborators. In addition, we may face competition from new entrants into
the field. We may not have the financial resources, technical expertise or marketing, distribution or support capabilities to compete
successfully in the future. The effects of any such actions of our competitors may have a materially adverse effect on our business,
operating results and financial condition.
Cyber-attacks
or other failures in telecommunications or information technology systems and deficiency in our, or those of third parties upon which
we rely, cybersecurity could result in information theft, data corruption and significant disruption of our business operations.
In
the ordinary course of business, we and the third parties upon which we rely and may process proprietary, confidential, and sensitive
data, including personal data (such as health-related data), intellectual property, and trade secrets (collectively, sensitive information).
Cyber-attacks, malicious internet-based activity, online and offline fraud, and other similar activities threaten the confidentiality,
integrity, and availability of our sensitive information and information technology systems, and those of the third parties upon which
we rely. Such threats are prevalent and continue to rise, are increasingly difficult to detect, and come from a variety of sources, including
traditional computer “hackers,” threat actors, “hacktivists,” organized criminal threat actors, personnel (such
as through theft or misuse), sophisticated nation states, and nation-state-supported actors.
Some
actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical
reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the
third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, that could
materially disrupt our systems and operations, supply chain, and ability conduct our research and development programs and our clinical
trials. We and the third parties upon which we rely may be subject to a variety of evolving threats, including but not limited to social-engineering
attacks (including through phishing attacks and deep fakes, which may be increasingly more difficult to identify as fake), malicious
code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks,
credential stuffing attacks, credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software
bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, telecommunications
failures, and other similar threats. In particular, severe ransomware attacks are becoming increasingly prevalent and severe and can
lead to significant interruptions in our operations, disruption of clinical trials, loss of sensitive data (including data related to
clinical trials), loss of income, reputational harm, and diversion of funds. Extortion payments may alleviate the negative impact of
a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting
such payments. Remote work has become more common and has increased risks to our information technology systems and data, as more of
our employees utilize network connections, computers, and devices outside of our premises or network, including working at home, while
in transit and in public locations. Future or past business transactions (such as acquisitions or integrations) could expose us to additional
cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated
entities’ systems and technologies.
In
addition, our reliance on third-party service providers could introduce new cybersecurity risks and vulnerabilities, including supply-chain
attacks, and other threats to our business operations. We may rely on third- party service providers and technologies to operate critical
business systems to process sensitive information in a variety of contexts, including, without limitation, third-party providers of information
technology infrastructure, cloud-based infrastructure, encryption and authentication technology, employee email, content delivery to
customers, CROs for managing clinical trial data, and other functions. We may also rely on third-party service providers to provide other
products, services, parts, or otherwise operate our business. Our ability to monitor these third parties’ information security
practices is limited, and these third parties may not have adequate information security measures in place. If our third-party service
providers experience a security incident or other interruption, we could experience adverse consequences. While we may be entitled to
damages if our third-party service providers fail to satisfy their privacy or security-related obligations to us, the liability of such
third party may be limited such that any award may be insufficient to cover our damages, or we may be unable to recover any such award.
In addition, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties’ infrastructure
in our supply chain or our third-party partners’ supply chains have not been compromised.
We
may expend significant resources or modify certain of our business activities (which could include our clinical trial activities) to
try to protect against security incidents. Certain data privacy and security obligations may require us to implement and maintain specific
security measures, industry-standard or reasonable security measures to protect our information technology systems and sensitive information.
While
we have established physical, electronic and organizational security measures designed to safeguard and secure our systems against security
incidents, there can be no assurance that these measures will be effective. We take steps designed to detect, mitigate and remediate
vulnerabilities in our information technology systems (such as our hardware and/or software, including that of third parties upon which
we rely). We may not, however, detect and remediate all such vulnerabilities including on a timely basis. Further, we may experience
delays in developing and deploying remedial measures and patches designed to address identified vulnerabilities.
Vulnerabilities
could be exploited and result in a security incident. Any of the previously identified or similar threats could cause a security incident
or other interruption that could result in unauthorized, unlawful, or accidental acquisition, modification, destruction, loss, alteration,
encryption, disclosure of, or access to our sensitive information or our information technology systems, or those of the third parties
upon whom we rely. A security incident or other interruption could disrupt our ability (and that of third parties upon whom we rely)
to provide our products. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result
in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.
Applicable
data privacy and security obligations may require us to notify relevant stakeholders of security incidents. Such disclosures are costly,
and the disclosure or the failure to comply with such requirements could lead to adverse consequences. If we (or a third party upon whom
we rely) experience a security incident or are perceived to have experienced a security incident, we may experience adverse consequences.
These consequences may include: government enforcement actions (for example, investigations, fines, penalties, audits, and inspections);
additional reporting requirements and/or oversight; restrictions on processing sensitive information (including personal data);
litigation (including class-action claims); indemnification obligations; negative publicity; reputational harm; monetary
fund diversions; interruptions in our operations (including availability of data); financial loss; and other similar harms.
Our
contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in
our contracts are sufficient to protect us from liabilities, damages, or claims related to our data privacy and security obligations.
We cannot be sure that our insurance coverage will be adequate or sufficient to protect us from or to mitigate liabilities arising out
of our privacy and security practices, that such coverage will continue to be available on commercially reasonable terms or at all, or
that such coverage will pay future claims.
In
addition to experiencing a security incident, third parties may gather, collect, or infer sensitive information about us from public
sources, data brokers, or other means that reveals competitively sensitive details about our organization and could be used to undermine
our competitive advantage or market position.
If
we do not successfully manage our growth, our business goals may not be achieved.
To
manage growth, we will be required to continue to improve existing, and implement additional, operational and financial systems, procedures
and controls, and hire, train and manage additional employees. Our current and planned personnel, systems, procedures and controls may
not be adequate to support our anticipated growth and we may not be able to hire, train, retain, motivate and manage required personnel.
Competition for qualified personnel in the biotechnology and regenerative medicine area is intense, and we operate or plan to operate
in geographic locations where labor markets are particularly competitive, including Boston, Massachusetts, where demand for personnel
with these skills is extremely high and is likely to remain high. As a result, competition for qualified personnel is intense and the
process of hiring suitably qualified personnel is often lengthy and expensive, and may become more expensive in the future. If we are
unable to hire and retain a sufficient number of qualified employees or otherwise manage our growth effectively, our ability to conduct
and expand our business could be seriously reduced.
Risks
Associated with Clinical Trials and Pre-Clinical Development
The
results of our clinical trials or pre-clinical development efforts may not support our product claims or may result in the discovery
of adverse side effects.
Even
if our pre-clinical development efforts or clinical trials are completed as planned, we cannot be certain that their results will support
our product claims or that the U.S. Food and Drug Administration, or FDA, foreign regulatory authorities or notified bodies will agree
with our conclusions regarding them. Although we have obtained some positive results from the use of our scaffolds and bioreactors for
esophageal and trachea implants performed to date, we also discovered that our first-generation trachea product design encountered certain
body response issues that we have sought to resolve with our ongoing development of our implant design. We cannot be certain that our
implant design or any future modifications or improvements with respect thereto will support our claims, and any such developments may
result in the discovery of further adverse side effects. We also may not see positive results when our product candidates undergo clinical
testing in humans in the future. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials
will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies.
Our pre-clinical development efforts and any clinical trial process may fail to demonstrate that our product candidates are safe and
effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Also,
patients receiving surgeries using our product candidates under compassionate use or in clinical trials may experience significant adverse
events following the surgeries, including serious health complications or death, which may or may not be related to materials provided
by us. In 2017, our esophageal implant candidate was used in a human surgery at Mayo Clinic via an FDA-approved single-use expanded access
application. In 2013 and 2014 we had provided a previous generation trachea scaffold device that was used in implants in human patients
under compassionate use. To date, we believe that at least four of the six patients who received those tracheal implants have died. While
we believe that none of those patients died because of a failure of the applicable device, these and any other such events have and may
cause or contribute to the delay or termination of our clinical trials or pre-clinical development efforts. Any delay or termination
of our pre-clinical development efforts or clinical trials will delay the filing of our product submissions and, ultimately, our ability
to commercialize our products and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse
side effects that are not currently part of the product candidate’s profile.
Regulatory
approval delays due to COVID-19
COVID-19
may impede clinical trials and slow down regulatory actions. It could adversely affect the entire clinical trial spectrum from enrollment
to data analysis. Assuming patients enroll, clinical trials may face disruptions to protocol schedules for treatment and follow-up visits.
Reports from Europe have noted overwhelmed facilities where all non-critical visits have been postponed or canceled. Many U.S. hospitals
have followed suit to limit exposure and allow for care of COVID-19 patients. Deviations from trial protocols could present challenges
when it comes time to analyze the related data set. Some clinics may stop allowing clinical trial monitors on site. Without reconciling
the data, we may be unable to “lock” the trial database, an essential step that precedes the analysis of the data.
We
rely on regular interaction and guidance from the FDA and other regional/country regulatory authorities/agencies to plan research and
development activities across all stages. Due to the COVID-19 pandemic, the FDA and worldwide regulatory authorities have a great deal
of resources dedicated to COVID-19 related matters, resulting in disruption in their ability to fully support the regulatory clearance/approval
processes. As resources continue to be diverted, regulatory clearances/approvals may continue to be delayed, until the pandemic is under
control. Therefore, delays with approvals, clearances, inspections, and meetings that are currently being experienced may continue for
the foreseeable future. Postponement of these interactions could delay us from bringing our product candidates to market.
Clinical
trials necessary to support a biological product license or other marketing authorization for our product candidates will be expensive
and will require the enrollment of sufficient patients to adequately demonstrate safety and efficacy for the product’s target populations.
Suitable patients may be difficult to identify and recruit. Delays or failures in our clinical trials will prevent us from commercializing
any products and will adversely affect our business, operating results and prospects.
In
the U.S., initiating and completing clinical trials necessary to support Biological License Applications, or BLAs, will be time consuming,
expensive and the outcome uncertain. Moreover, the FDA may not agree that clinical trial results support an application for the indications
sought in the application for the product. In other jurisdictions such as the E.U., the conduct of extensive and expensive clinical trials
may also be required in order to demonstrate the quality, safety and efficacy of our product candidates, depending on each specific product
candidate, the claims being studied, and the target condition or disease. The outcome of these clinical trials, which can be expensive
and are heavily regulated, will also be uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future
results, and any product candidate we advance into clinical trials following initial positive results in early clinical trials may not
have favorable results in later clinical trials.
Conducting
successful clinical trials will require the enrollment of a sufficient number of patients to support each trial’s claims, and suitable
patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and
follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the attractiveness
of, or the discomfort and risks associated with, the treatments received by enrolled subjects, the availability of appropriate clinical
trial investigators, support staff, and proximity of patients to clinical sites and ability to comply with the eligibility and exclusion
criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our
clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety
and effectiveness of our product candidates, or if they determine that the treatments received under the trial protocols are not attractive
or involve unacceptable risks or discomfort. Also, patients may not participate in our clinical trials if they choose to participate
in contemporaneous clinical trials of competitive products. In addition, patients participating in clinical trials may die before completion
of the trial or suffer adverse medical events unrelated to investigational products.
Development
of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required and we may not adequately develop such
protocols to support clearance and approval. Further, the FDA and foreign regulatory authorities may require us to submit data on a greater
number of patients than we originally anticipated and/or for a longer follow-up period or change the data collection requirements or
data analysis applicable to our clinical trials. Delays in patient enrollment or failure of patients to continue to participate in a
clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our products or result in
the failure of the clinical trial. In addition, despite considerable time and expense invested in our clinical trials, the FDA and foreign
regulatory authorities may not consider our data adequate to demonstrate safety and efficacy. Although FDA regulations allow submission
of data from clinical trials outside the U.S., there can be no assurance that such data will be accepted or that the FDA will not apply
closer scrutiny to such data. Increased costs and delays necessary to generate appropriate data, or failures in clinical trials could
adversely affect our business, operating results and prospects.
If
the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually-required
or expected, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We
do not have the ability to independently conduct our pre-clinical and clinical trials for our product candidates and we must rely on
third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct,
or assist us in conducting, such trials, including data collection and analysis. We do not have direct control over such third parties’
personnel or operations. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet
expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised
due to the failure to adhere to our clinical protocols or any regulatory requirements, or for other reasons, our pre-clinical development
activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to seek or obtain regulatory
approval for, or successfully commercialize, our product candidates on a timely basis, if at all. Our business, operating results and
prospects may also be adversely affected. Furthermore, any third-party clinical trial investigators pertaining to our product candidates
may be delayed in conducting our clinical trials for reasons outside of their control.
Risks
Associated with Regulatory Approvals
If
we fail to obtain, or experience significant delays in obtaining, regulatory approvals in the U.S., China or the E.U. for our products,
including those for the esophagus and airways, or are unable to maintain such clearances or approvals for our products, our ability to
commercially distribute and market these products would be adversely impacted.
We
currently do not have regulatory approval to market any of our implant product candidates, including those for the esophagus, colon,
and uterus. Our product candidates are subject to rigorous regulation by the FDA, and numerous other federal and state governmental authorities
in the U.S., as well as foreign governmental authorities. In the U.S., the FDA permits commercial distribution of new medical products
only after approval of a Premarket Approval, or PMA, New Drug Application, or NDA, or BLA, unless the product is specifically exempt
from those requirements. A PMA, NDA or BLA must be supported by extensive data, including, but not limited to, technical, pre-clinical,
clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the product
for its intended use. There are similar approval processes in China, the E.U. and other foreign jurisdictions. Our failure to receive
or obtain such clearances or approvals on a timely basis or at all would have an adverse effect on our results of operations.
The
first bioengineered trachea implant approved in the U.S. using our first-generation trachea scaffold in an implant was approved under
the IND pathway through the FDA’s Center for Biologics Evaluation and Research, or CBER, for a single compassionate use. Such initial
U.S. surgery was led by Professor Paolo Macchiarini, M.D., a surgeon pioneering tracheal replacement techniques. Dr. Macchiarini was
not employed or affiliated with our company, and we did not pay him any compensation or consulting fees. In June 2014, we ceased support
of any human surgeries with Dr. Macchiarini. Since the time we withdrew from involvement with Dr. Macchiarini, allegations that Dr. Macchiarini
had failed to obtain informed consent and accurately report patient conditions, among other things, for surgeries performed at the Karolinska
Institutet in Stockholm, Sweden, were made public.
The
Karolinska Institutet investigated the allegations and concluded that while in some instances Dr. Macchiarini did act without due care,
his actions did not qualify as scientific misconduct. Subsequent to this investigation, further negative publicity and claims continued
to be released questioning the conduct of Dr. Macchiarini, the Karolinska Institutet, the Krasnodar Regional Hospital in Krasnodar, Russia
as well as our company relating to surgeries performed by Dr. Macchiarini and other surgeons at such facilities. In February 2015, the
Karolinska Institutet announced that it would conduct an additional investigation into the allegations made about Dr. Macchiarini and
the Karolinska Institutet’s response and actions in the earlier investigation. In March 2015, the Karolinska Institutet announced
that it was terminating Dr. Macchiarini’s employment, and in December 2016 the Karolinska Institutet found Dr. Macchiarini, along
with three co-authors, guilty of scientific misconduct. In May 2022, Dr. Macchiarini was tried in Solna District Court in Sweden for
aggravated assault against three patients treated at the Karolinska University Hospital. On June 16, 2022, Dr. Macchiarini was acquitted
in two of these cases and in the third was found guilty of causing bodily harm to the patient and was given a suspended sentence for
two years. These allegations, the results of the investigation and any further actions that may be taken in connection with these matters,
have and may continue to harm the perception of our product candidates or company and make it difficult to recruit patients for any clinical
trials, which could have a material adverse effect on our business, financial condition or results of operations.
The
FDA has informed us that our esophageal implant would be viewed by the FDA as a combination product comprised of a biologic, or cells,
and a medical device component. Nevertheless, we cannot be certain how the FDA will regulate our products. The FDA may require us to
obtain marketing clearance and approval from multiple FDA centers. The review of combination products is often more complex and more
time consuming than the review of products under the jurisdiction of only one center within the FDA.
While
the FDA has informed us that our esophageal implant would be regulated by the FDA as a combination product, we cannot be certain that
any of our other products would also be regulated by the FDA as a combination product. For a combination product, the Office of Combination
Products, or OCP, within FDA can determine which center or centers within the FDA will review the product and under what legal authority
the product will be reviewed. Generally, the center within the FDA that has the primary role in regulating a combination product is determined
based on the primary mode of action of the product. Generally, if the primary mode of action is as a device, the FDA’s Center for
Devices and Radiological Health, or CDRH, takes the lead. Alternatively, if the primary mode of action is cellular, then the CBER takes
the lead. On October 18, 2016, we also received written confirmation from the CBER that the FDA intends to regulate our esophageal implant
as a combination product under the primary jurisdiction of CBER. We further understand that CBER may choose to consult or collaborate
with CDRH with respect to the characteristics of the synthetic scaffold component of our product based on CBER’s determination
of need for such assistance.
The
process of obtaining FDA marketing approval is lengthy, expensive, and uncertain, and we cannot be certain that our product candidates,
including product candidates pertaining to the esophagus, airways, or otherwise, will be cleared or approved in a timely fashion, or
at all. In addition, the review of combination products is often more complex and can be more time consuming than the review of a product
under the jurisdiction of only one center within the FDA.
We
cannot be certain that the FDA will not elect to have our combination product candidates reviewed and regulated by only one FDA center
and/or different legal authority, in which case the path to regulatory approval would be different and could be more lengthy and costly.
If
the FDA does not approve or clear our products in a timely fashion, or at all, our business, financial condition or operations will be
adversely affected.
In
the E.U., our esophagus product candidate will likely be regulated as a combined advanced therapy medicinal product and our other product
candidates, including for the colon and uterus, may also be viewed as advanced therapy medicinal products, which could delay approvals
and clearances and increase costs of obtaining such approvals and clearances.
On
May 28, 2014, we received notice from the European Medicines Agency, or EMA, that our first-generation trachea product candidate
would be regulated as a combined advanced therapy medicinal product. While we have not had any formal interaction with the EMA with respect
to our esophageal implant, we believe that such implant technology would likely be regulated as a combined advanced therapy medicinal
product. In the event of such classification, it would be necessary to seek a marketing authorization for these products granted by the
European Commission before being marketed in the E.U.
Other
products we may develop, including any products pertaining to the airways or otherwise, may similarly be regulated as advanced therapy
medicinal products or combined advanced therapy medicinal products. The regulatory procedures leading to marketing approval of our products
vary among jurisdictions and can involve substantial additional testing. Compliance with the FDA requirements does not ensure clearance
or approval in other jurisdictions, and the ability to legally market our products in any one foreign country does not ensure clearance,
or approval by regulatory authorities in other foreign jurisdictions. The foreign regulatory process leading to the marketing of the
products may include all of the risks associated with obtaining FDA approval in addition to other risks. In addition, the time required
to comply with foreign regulations and market products may differ from that required to obtain FDA approval, and we may not obtain foreign
approval or clearance on a timely basis, if at all.
Risk
Associated with Product Marketing
Even
if our products are cleared or approved by regulatory authorities, if we or our suppliers fail to comply with ongoing FDA or other foreign
regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions
or withdrawal from the market.
Any
product for which we obtain clearance or approval in the U.S., China, or Europe, and the manufacturing processes, reporting requirements,
post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and
periodic inspections by the FDA and other domestic and foreign regulatory authorities or notified bodies. In particular, we and our suppliers
are required to comply with the FDA’s Quality System Regulations, or QSR, and current Good Manufacturing Practices, or cGMP, for
our medical products, and International Standards Organization, or ISO, regulations for the manufacture of our products and other regulations
which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage
and shipping of any product for which we obtain clearance or approval. Manufacturing may also be subject to controls by the FDA for parts
of the system or combination products that the FDA may find are controlled by the biologics regulations. Equivalent regulatory obligations
apply in foreign jurisdictions. Regulatory authorities, such as the FDA, China’s National Medical Products Administration, the
competent authorities of the E.U. Member States, the EMA and notified bodies, enforce the QSR, cGMP and other applicable regulations
in the U.S. and in foreign jurisdictions through periodic inspections. The failure by us or one of our suppliers to comply with applicable
statutes and regulations administered by the FDA and other regulatory authorities or notified bodies in the U.S. or in foreign jurisdictions,
or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among
other things, any of the following enforcement actions:
| |
● |
untitled
letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
● |
unanticipated
expenditures to address or defend such actions; |
| |
● |
customer
notifications for repair, replacement, or refunds; |
| |
● |
recall,
detention or seizure of our products; |
| |
● |
operating
restrictions or partial suspension or total shutdown of production; |
| |
● |
withdrawing
BLA or NDA approvals that have already been granted; |
| |
● |
withdrawal
of the marketing authorization granted by the European Commission or delay in obtaining such marketing authorization; |
| |
● |
withdrawal
of the CE Certificates of Conformity granted by the notified body or delay in obtaining these certificates; |
| |
● |
refusal
to grant export approval for our products; and |
| |
● |
criminal
prosecution. |
The
occurrence of any of these events could have a material adverse effect on our business, financial condition or results of operations.
Post-market
enforcement actions can generate adverse commercial consequences.
Even
if regulatory approval of a product is granted, such clearance or approval may be subject to limitations on the intended uses for which
the product may be marketed and reduce our potential to successfully commercialize the product and generate revenue from the product.
If the FDA or a foreign regulatory authority determines that our promotional materials, labeling, training or other marketing or educational
activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials
or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might
take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result
in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In addition,
we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and
we must comply with medical products reporting requirements, including the reporting of adverse events and malfunctions related to our
products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events
of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result
in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary
or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines,
suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely
affect our business, operating results and prospects.
Economic,
political and other risks associated with our international operations could adversely affect our revenues and international growth prospects.
The
Company started selling longevity supplements through Longevity Products in the third quarter of 2023. These products are marketed
to the general public and initially targeted at consumers in the Great China Region through eCommerce (online sales).
Longevity Products plans to include a broad
range of products focused on personal healthcare including longevity dietary supplements. Our international operations are subject
to a number of risks inherent to operating in foreign countries, and any expansion of our international operations will amplify the
effects of these risks, which include, among others:
| ● |
political and economic instability of foreign markets; |
| ● |
foreign governments’ restrictive trade policies or the
impact of trade tensions amongst nations; |
| ● |
inconsistent product regulation or sudden policy changes by
foreign agencies or governments; |
| ● |
the imposition of, or increase in, duties, taxes, government
royalties or non-tariff trade barriers; |
| ● |
difficulty in collecting international accounts receivable
and potentially longer payment cycles; |
| ● |
difficulty of enforcing contractual obligations of foreign
partners; |
| ● |
increased costs in maintaining international marketing efforts; |
| ● |
problems entering international markets with different cultural
bases and consumer preferences; |
| ● |
compliance with foreign regulatory requirements such as the
General Data Protection Regulation (GDPR), domestic laws and regulations applicable to international operations, such as the Foreign
Corrupt Practices Act and regulations promulgated by the Office of Foreign Asset Control, as well as regulatory laws, regulations and
restrictions that may impact or target dietary supplement retailers and manufacturers; |
| ● |
fluctuations in foreign currency exchange rates; and |
| ● |
operating in new, developing or other markets in which there
are significant uncertainties regarding the interpretation, application and enforceability of laws and regulations relating to contract
and intellectual property rights. |
Any
of these risks could have a material adverse effect on our international operations and our growth strategy.
Additionally,
if the opportunity arises, we may expand our operations into new and high-growth international markets. However, there is no assurance
that we will expand our operations in such markets in our desired time frame. To expand our operations into new international markets,
we may enter into business combination transactions, make acquisitions or enter into strategic partnerships, joint ventures or alliances,
any of which may be material. We may enter into these transactions to acquire other businesses or products to expand our products or
take advantage of new developments and potential changes in the industry. Our lack of experience operating in new international markets
and our lack of familiarity with local economic, political and regulatory systems could prevent us from achieving the results that we
expect on our anticipated time frame or at all. If we are unsuccessful in expanding into new or high-growth international markets, it
could adversely affect our operating results and financial condition.
Additionally,
our business is increasingly exposed to operational risks in China. These include, among others, changes in economic conditions (including
consumer spending, unemployment levels and wage and commodity inflation), consumer preferences, the regulatory environment, and tax laws
and regulations, as well as increased media scrutiny, fluctuations in foreign exchange rates, increased restrictions or tariffs on imported
supplies as a result of trade disputes and increased competition. Any significant or prolonged deterioration in U.S.-China relations
could adversely affect our operations in China if Chinese consumers reduce the frequency of their purchases of our products. Chinese
law regulates our business conducted within China. In addition, if we are unable to enforce our intellectual property or contract rights
in China, it could result in an interruption in the operation of our brands, which could negatively impact our financial results. If
our business is harmed or development of our Chinese operations is slowed in China due to any of these factors, it could negatively impact
our overall financial results or our growth prospects.
Risks
Relating to Our Common Stock
Our
principal stockholders hold a significant percentage of our voting power and will be able to exert significant control over us.
The
stockholders who purchased shares of our common stock and related warrants pursuant to a Securities Purchase Agreement dated December
27, 2017 collectively hold shares of common stock that represent approximately 32% of all outstanding voting power, and as such may significantly
influence the results of matters voted on by our shareholders. The interests of these stockholders may conflict with your interests.
This significant concentration of share ownership may adversely affect the trading price for our common stock because investors may perceive
disadvantages in owning stock in companies with controlling stockholders.
A
trading market that will provide you with adequate liquidity may not develop for our common stock.
The
current public market for our common stock has limited trading volume and liquidity. We cannot predict the extent to which investor interest
in our company will lead to the development of a more active trading market in our common stock, or how liquid that market might be.
Our
revenues, operating results and cash flows may fluctuate in future periods and we may fail to meet investor expectations, which may cause
the price of our common stock to decline.
Variations
in our quarterly and year-end operating results are difficult to predict and may fluctuate significantly from period to period. If our
revenues or operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline
substantially, which could have a material adverse effect on our ability to raise additional capital, to use our stock as consideration
for future acquisitions or for compensation of our employees. In addition to the other factors discussed under these “Risk Factors,”
specific factors that may cause fluctuations in our operating results include:
| |
● |
demand
and pricing for our products; |
| |
● |
government
or private healthcare reimbursement policies; |
| |
● |
adverse
events or publicity related to our products, our research or investigations, or our collaborators or other partners; |
| |
● |
physician
and patient acceptance of any of our current or future products; |
| |
● |
manufacturing
stoppages or delays; |
| |
● |
introduction
of competing products or technologies; |
| |
● |
our
operating expenses which fluctuate due to growth of our business; and |
| |
● |
timing
and size of any new product or technology acquisitions we may complete. |
Substantial
sales of common stock have and may continue to occur, or may be anticipated, which have and could continue to cause our stock price to
decline.
We
expect that we will seek to raise additional capital from time to time in the future, which may involve the issuance of additional shares
of common stock, or securities convertible into or exercisable for common stock. The purchasers of the shares of common stock and warrants
to purchase shares of common stock from our public offerings and private placements may sell significant quantities of our common stock
in the market, which may cause a decline in the price of our common stock. Further, we cannot predict the effect, if any, that any additional
market sales of common stock, or anticipation of such sales, or the availability of those shares of common stock for sale will have on
the market price of our common stock. Any future sales of significant amounts of our common stock, or the perception in the market that
this will occur, may result in a decline in the price of our common stock.
Your
percentage ownership will be diluted in the future.
Your
percentage ownership will be diluted in the future because of equity awards that we expect will be granted to our directors, officers
and employees, as well as shares of common stock, or securities convertible into common stock, we issue in connection with future capital
raising or strategic transactions. Our Amended and Restated Equity Incentive Plan provides for the grant of equity-based awards, including
restricted stock, restricted stock units, stock options, stock appreciation rights and other equity-based awards to our directors, officers
and other employees, advisors and consultants. The issuance of any shares of our stock would dilute the proportionate ownership and voting
power of existing security holders.
Provisions
of Delaware law, of our amended and restated charter and amended and restated bylaws may make a takeover more difficult, which could
cause our stock price to decline.
Provisions
in our amended and restated certificate of incorporation and amended and restated bylaws and in the Delaware corporate law may make it
difficult and expensive for a third party to pursue a tender offer, change in control or takeover attempt, which is opposed by management
and the Board of Directors. Public stockholders who might desire to participate in such a transaction may not have an opportunity to
do so. We have a staggered Board of Directors that makes it difficult for stockholders to change the composition of the Board of Directors
in any one year. Any removal of directors will require a super-majority vote of the holders of at least 75% of the outstanding shares
entitled to be cast on the election of directors which may discourage a third party from making a tender offer or otherwise attempting
to obtain control of us. These anti-takeover provisions could substantially impede the ability of public stockholders to change our management
and Board of Directors. Such provisions may also limit the price that investors might be willing to pay for shares of our common stock
in the future.
The
market price of our shares may fluctuate widely.
The
market price of our common stock may fluctuate widely, depending upon many factors, some of which may be beyond our control, including:
| ● |
the
success and costs of preclinical and clinical testing and obtaining regulatory approvals or clearances for our products; |
| ● |
the
success or failure of surgeries and procedures involving the use our products; |
| ● |
a
shift in our investor base; |
| ● |
our
quarterly or annual results of operations, or those of other companies in our industry; |
| ● |
actual
or anticipated fluctuations in our operating results due to factors related to our business; |
| ● |
changes
in accounting standards, policies, guidance, interpretations or principles; |
| ● |
announcements
by us or our competitors of significant acquisitions, dispositions or intellectual property developments or issuances; |
| ● |
the
failure of securities analysts to cover our common stock; |
| ● |
changes
in earnings estimates by securities analysts or our ability to meet those estimates; |
| ● |
the
operating and stock price performance of other comparable companies; our issuance of equity, debt or other financing instruments; |
| ● |
overall
market fluctuations; and |
| ● |
general
macroeconomic conditions. |
Stock
markets in general have experienced volatility that has often been unrelated to the operating performance of a particular company. These
broad market fluctuations may adversely affect the trading price of our common stock.
Any
issuance of preferred stock in the future may dilute the rights of our common stockholders.
Our
Board of Directors has the authority to issue up to 2,000,000 shares of preferred stock and to determine the price, privileges and other
terms of these shares. Our Board of Directors is empowered to exercise this authority without any further approval of stockholders. The
rights of the holders of common stock may be adversely affected by the rights of future holders of preferred stock.
We
have in the past issued, and we may at any time in the future issue, additional shares of authorized preferred stock. For example,
in our December 2017 private placement transaction, we authorized 12,000 shares of Series D convertible preferred stock, of which we
issued 3,108 shares, all of which have been converted into shares of common stock, and in June 2022 we also issued 4,000 shares of
Series E convertible preferred stock, and additional shares of Series E convertible preferred stock thereafter in relation to
dividends on such Series E convertible preferred stock. The Company issued an aggregate of 180 and 77 shares of Series E convertible
preferred stock relating to accrued dividends during the years ended December 31, 2022 and 2023, respectively. All shares of Series E convertible
preferred stock have been converted into shares of common stock as of December 31, 2023.
We
do not intend to pay cash dividends on our common stock.
Currently,
we do not anticipate paying any cash dividends to holders of our common stock. As a result, capital appreciation, if any, of our common
stock will be a stockholder’s sole source of gain.
Our
common stock has been delisted on the NASDAQ Capital Market, which may negatively impact the trading price of our common stock and the
levels of liquidity available to our stockholders.
Our
common stock was suspended from trading on the NASDAQ Capital Market, prior to the opening of the market on October 6, 2017 and began
quotation on the OTCQB Venture Market on that date, retaining the symbol “BSTG”. On December 7, 2017, the NASDAQ Capital
Market filed a Form 25-NSE with the SEC to complete the delisting process. The trading of our common stock on the OTCQB Venture Market
rather than The NASDAQ Capital Market may negatively impact the trading price of our common stock and the levels of liquidity available
to our stockholders.
Upon
such delisting, our common stock became subject to the regulations of the SEC relating to the market for penny stocks. A penny stock
is any equity security not traded on a national securities exchange that has a market price of less than $5.00 per share. The regulations
applicable to penny stocks may severely affect the market liquidity for our common stock and could limit the ability of shareholders
to sell securities in the secondary market. Accordingly, investors in our common stock may find it more difficult to dispose of or obtain
accurate quotations as to the market value of our common stock, and there can be no assurance that our common stock will continue to
be eligible for trading or quotation on the OTCQB Venture Market or any other alternative exchanges or markets.
The
delisting of our common stock from the NASDAQ Capital Market may adversely affect our ability to raise additional financing through public
or private sales of equity securities, may significantly affect the ability of investors to trade our securities, and may negatively
affect the value and liquidity of our common stock. Such delisting may also have other negative results, including the potential loss
of confidence by employees, the loss of institutional investor interest and fewer business development opportunities. Furthermore, because
of the limited market and low volume of trading in our common stock that could occur, the share price of our common stock could more
likely be affected by broad market fluctuations, general market conditions, fluctuations in our operating results, changes in the market’s
perception of our business, and announcements made by us, our competitors, parties with whom we have business relationships or third
parties.
If
securities or industry analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business
or our market, our stock price and trading volume could decline.
The
trading market for our common stock will be influenced by the research and reports that securities or industry analysts publish about
us and our business. Securities or industry analysts may elect not to provide coverage of our common stock, and such lack of coverage
may adversely affect the market price of our common Stock. In the event we do not secure additional securities or industry analyst coverage,
we will not have any control over the analysts or the content and opinions included in their reports. The price of our stock could decline
if one or more securities or industry analysts downgrade our stock or issue other unfavorable commentary or research. If one or more
securities or industry analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our stock could
decrease, which in turn could cause our stock price or trading volume to decline.
General
Risk Factors
Impact
of COVID-19, Supply Chain Disruptions and Other Matters
The
impact of the COVID-19 outbreak has subsided substantially in the U.S. but continues to result in reduced activity levels outside of
the U.S., such as continued restrictions on travel and business operations and advising or requiring individuals to limit or forego their
time outside of their homes or places of business. In response to the global supply chain instability and inflationary cost increases,
we have taken action to minimize, as much as possible, any potential adverse impacts by working with our suppliers to monitor the availability
of raw material components (e.g., polymers and organic solvents), lead times, and freight carrier availability. We expect global supply
chain instability will continue to have an impact on our business, but to date that has not been material to our financial performance
or the development of our products. The consequences of the pandemic, global supply chain instability and inflationary cost increases
and their adverse impact to the global economy, continue to evolve. Accordingly, the significance of the future impact to our business,
financial condition and results of operations remains subject to significant uncertainty.
We
are subject to new U.S. foreign investment regulations, which may impose additional burdens on or may limit certain investors’
ability to purchase our common stock, potentially making our common stock less attractive to investors, and may also impact our ability
to generate revenues outside of the U.S.
In
October 2018, the U.S. Department of Treasury announced a pilot program to implement part of the Foreign Investment Risk Review Modernization Act of 2018 (FIRRMA),
effective November 10, 2018.
The pilot program expands the jurisdiction of CFIUS to include certain direct or indirect foreign investments in a defined category of
U.S. companies, which may include companies such as Harvard Apparatus Regenerative Technology in the biotechnology industry. Among other
things, FIRRMA empowers CFIUS to require certain foreign investors to make mandatory filings and permits CFIUS to charge filing fees
related to such filings. Such filings are subject to review by CFIUS. Any such restrictions on the ability to purchase shares of our
common stock may have the effect of delaying or deterring any particular investment and could also affect the price that some investors
are willing to pay for our common stock. In addition, such restrictions could also limit the opportunity for our stockholders to receive
a premium for their shares of our common stock in relation to any potential change in control.
We
intend to generate significant revenues outside the U.S., including in China and the E.U. Restrictions, such as those related to CFIUS,
not only affect foreign ownership and investments, but also the transfer or licensing of technology from the U.S. into certain foreign
markets, including China. Such restrictions, including to the extent they block strategic transactions that might otherwise be in stockholder’s
interests, may materially and adversely affect our ability to generate revenues in those foreign markets and the results of our operations.
If
we incur higher costs as a result of trade policies, treaties, government regulations or tariffs, it could have a materially adverse
effect on our business, financial condition or results of operations.
There
is currently significant uncertainty about the future relationship between the United States and China, including with respect to trade
policies, treaties, government regulations and tariffs. The current United States administration has called for substantial changes to
U.S. foreign trade policy including greater restrictions on international trade and significant increases in tariffs on goods imported
into the U.S. Under the current status, we do not expect that this tariff will significantly impact any Harvard Apparatus Regenerative
Technology products and thus the tariff should not have a materially adverse effect on our business, financial condition or results of
operations. We are unable to predict whether or when additional tariffs will be imposed or the impact of any such future tariff increases.
We
are exposed to a variety of risks relating to our potential international sales and operations, including fluctuations in exchange rates,
local economic conditions and delays in collection of accounts receivable.
We
intend to generate significant revenues outside the U.S. in multiple foreign currencies including Chinese Renminbi, Euros, British pounds,
and in U.S. dollar-denominated transactions conducted with customers who generate revenue in currencies other than the U.S. dollar. In
such instances, for those foreign customers who purchase our products in U.S. dollars, currency fluctuations between the U.S. dollar
and the currencies in which those customers do business may have a negative impact on the demand for our products in foreign countries
where the U.S. dollar has increased in value compared to the local currency.
Since
we may have vendors and customers outside the U.S. and we may generate revenues and incur operating expenses in multiple foreign currencies,
we will experience currency exchange risk with respect to any foreign currency-denominated revenues and expenses. We cannot predict the
consolidated effects of exchange rate fluctuations upon our future operating results because of the number of currencies involved, the
variability of currency exposure and the potential volatility of currency exchange rates. Our international activities subject us to
laws regarding sanctioned countries, entities and persons, customs, import-export, laws regarding transactions in foreign countries,
the U.S. Foreign Corrupt Practices Act and local anti-bribery and other laws regarding interactions with healthcare professionals. Among
other things, these laws restrict, and in some cases prohibit, U.S. companies from directly or indirectly selling goods, technology or
services to people or entities in certain countries. In addition, these laws require that we exercise care in structuring our sales and
marketing practices in foreign countries.
Local
economic conditions, legal, regulatory or political considerations, disruptions from strikes, the effectiveness of our sales representatives
and distributors, local competition and changes in local medical practice could also affect our sales to foreign markets. Relationships
with customers and effective terms of sale frequently vary by country, often with longer-term receivables than are typical in the U.S.
Comprehensive
tax reform legislation could adversely affect our business and financial condition.
In
December 2017, the U.S. government enacted the Tax Cuts and Jobs Act of 2017, or TCJA, which significantly reforms the Internal Revenue
Code of 1986, as amended. The TCJA, among other things, contains significant changes to corporate taxation, including reduction of the
corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, effective January 1, 2018; limitation of the tax deduction
for interest expense; limitation of the deduction for net operating losses and elimination of net operating loss carrybacks, in each
case, for losses arising in taxable years beginning after December 31, 2017 (though any such tax losses may be carried forward indefinitely);
and modifying or repealing many business deductions and credits, including reducing the business tax credit for certain clinical testing
expenses incurred in the testing of certain drugs for rare diseases or conditions generally referred to as “orphan drugs”.
The tax rate change resulted in (i) a reduction in the gross amount of our deferred tax assets recorded as of December 31, 2017, without
an impact on the net amount of our deferred tax assets, which are recorded with a full valuation allowance. We continue to examine the
impact this tax reform legislation may have on our business. However, the effect of the TCJA on us and our affiliates, whether adverse
or favorable, is uncertain and may not become evident for some period of time. We urge investors to consult with their legal and tax
advisers regarding the implications of the TCJA on an investment in our common stock.
Changes
in the European regulatory environment regarding privacy and data protection regulations could have a materially adverse impact on our
results of operations.
The
European Union, or E.U., has adopted a comprehensive overhaul of its data protection regime in the form of the General Data Protection
Regulation, or GDPR, which came into effect in May 2018. GDPR extends the scope of the existing E.U. data protection law to foreign companies
processing personal data of E.U. residents. The regulation imposes a strict data protection compliance regime with severe penalties of
4% of worldwide turnover or €20 million, whichever is greater, and includes new rights such as the right of erasure of personal
data. Although the GDPR will apply across the E.U., as has been the case under the current data protection regime, E.U. Member States
have some national derogations and local data protection authorities that will still have the ability to interpret the GDPR, which has
the potential to create inconsistencies on a country-by-country basis. Implementation of, and compliance with the GDPR could increase
our cost of doing business and/or force us to change our business practices in a manner adverse to our business. In addition, violations
of the GDPR may result in significant fines, penalties and damage to our brand and business which could, individually or in the aggregate,
materially harm our business and reputation.
Healthcare
legislative reform measures may have a materially adverse effect on our business and results of operations.
In
the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in
March 2010, the Affordable Care Act, or ACA, was passed, which substantially changes the way healthcare is financed by both governmental
and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA, among other things, subjects biological products
to potential competition by lower-cost biosimilars, addresses a new methodology by which rebates owed by manufacturers under the Medicaid
Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid
rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid
managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and creates a new
Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (70% commencing January 1, 2019) point-of-sale
discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition
for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Some of the provisions of the ACA have yet to be fully
implemented, while certain provisions have been subject to Judicial and Congressional challenges, as well as efforts by the Trump administration
to repeal or replace certain aspects of the ACA. Since January 2017, former President Trump signed two Executive Orders designed to delay
the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by
the ACA.
Concurrently,
Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive
repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. The TCJA includes
a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals
who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.”
Additionally, on January 22, 2018, former President Trump signed a continuing resolution on appropriations for fiscal year 2018 that
delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax, an annual fee on certain
high-cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and
the Medical Device Excise Tax, or MDET, on non-exempt medical devices. Since then, The Further Consolidated Appropriations Act, 2020
H.R. 1865, signed into law on December 20, 2019, repealed the MDET. Further, the Bipartisan Budget Act of 2018, or the BBA, among other
things, amends the ACA, effective January 1, 2019, to reduce the coverage gap in most Medicare drug plans, commonly referred to as the
“donut hole.” The effect that the ACA and its possible repeal and replacement may have on our business remains unclear.
Other
legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control
Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction,
tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach
required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate
reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent
legislative amendments to the statute, will remain in effect through 2027 unless additional congressional action is taken. On January
2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to
several types of providers.
Moreover,
payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, the Middle Class Tax
Relief and Job Creation Act of 2012 required that the Centers for Medicare & Medicaid Services, or CMS, the agency responsible for
administering the Medicare program, reduce the Medicare clinical laboratory fee schedule by 2% in 2013, which served as a base for 2014
and subsequent years. In addition, effective January 1, 2014, CMS also began bundling the Medicare payments for certain laboratory tests
ordered while a patient received services in a hospital outpatient setting. We expect that additional state and federal healthcare reform
measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare
products and services, which could result in reduced demand for any product candidate we develop or complementary diagnostics or companion
diagnostics or additional pricing pressures.
Additionally,
there has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices.
Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed
to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship
between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
Most
recently, the Inflation Reduction Act of 2022 included a number of significant drug pricing reforms, which include the establishment
of a drug price negotiation program within the U.S. Department of Health and Human Services that requires manufacturers to charge a negotiated
“maximum fair price” for certain selected drugs or pay an excise tax for noncompliance, the establishment of rebate payment
requirements on manufacturers under Medicare Parts B and D to penalize price increases that outpace inflation, and a redesign of the
Part D benefit, as part of which manufacturers are required to provide discounts on Part D drugs.
Any
of these regulatory changes and events could limit our ability to form collaborations and our ability to commercialize our products,
and if we fail to comply with any such new or modified regulations and requirements it could adversely affect our business, operating
results and prospects.
If
we fail to complete the required IRS forms for exemptions, make timely semi-monthly payments of collected excise taxes, or submit quarterly
reports as required by the MDET, we may be subject to penalties, such as Section 6656 penalties for any failure to make timely deposits.
Section
4191 of the Internal Revenue Code, enacted by Section 1405 of the Health Care and Education Reconciliation Act of 2010, Public Law 111-152
(124 Stat. 1029 (2010)), in conjunction with the Patient Protection and the ACA, Public Law 111-148 (124 Stat. 119 (2010)), imposed as
of January 1, 2013, an excise tax on the sale of certain medical devices. The MDET imposed by Section 4191 is 2.3% of the price for which
a taxable medical device is sold within the U.S.
We
are a smaller reporting company and the reduced disclosure requirements applicable to smaller reporting companies may make our common
stock less attractive to investors.
We
are a smaller reporting company, or SRC, and a non-accelerated filer, which allows us to take advantage of exemptions from various reporting
requirements that are applicable to other public companies that are not SRCs or non-accelerated filers, including not being required
to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, reduced disclosure
obligations, including disclosures regarding executive compensation, in our Annual Report and our periodic reports and proxy statements
and providing only two years of audited financial statements in our Annual Report and our periodic reports. We will remain an SRC until
(a) the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed
second fiscal quarter exceeds $250 million or (b) in the event we have over $100 million in annual revenues, and the aggregate market
value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed second fiscal quarter
exceeds $700 million. We cannot predict whether investors will find our common stock less attractive if we rely on certain or all of
these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for
our common stock and our stock price may be more volatile and may decline.
We
incur increased costs as a result of operating as a public company, and our management is required to devote substantial time to new
compliance initiatives and corporate governance practices.
As
a public company, we incur significant legal, accounting, and other expenses that we did not incur as a private company. These costs
generally increase for a company whose shares are listed on the NYSE American or Nasdaq Capital Market as compared to the costs for a
company for whose shares are quoted on the OTCQB Venture Market. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer
Protection Act, FINRA rules and other applicable securities rules and regulations impose various requirements on public companies, including
establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other
personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase
our legal and financial compliance costs and make some activities more time-consuming and costly.
We
continue to evaluate these rules and regulations and cannot predict or estimate the amount of additional costs we may incur or the timing
of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity,
and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies.
This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure
and governance practices.
We
will likely in the future have assets held at financial institutions that may exceed the insurance coverage offered by the Federal Deposit
Insurance Corporation (“FDIC”), the loss of which would have a severe negative affect on our operations and liquidity.
We
currently have the majority of our cash and cash equivalents held in deposit at East West Bank. While the amounts held in the deposit
accounts as of December 31, 2023 were less than the insurance coverage offered by the FDIC, in the future, we will likely maintain our cash
assets at financial institutions in the U.S. in amounts that may be in excess of the FDIC insurance limit of $250,000. Actual events
involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional
counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors
about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems.
For example, on March 10, 2023, Silicon Valley Bank, or SVB, was closed by the California Department of Financial Protection and Innovation,
which appointed the FDIC as receiver. In the event of a failure or liquidity issue of or at any of these financial institutions where
we maintain our deposits or other assets, we may incur a loss, and to the extent such loss exceeds the FDIC insurance limitation it could
have a material adverse effect upon our liquidity, financial condition and our results of operations.
Item
1B. Unresolved Staff Comments.
None.
Item
1C. Cybersecurity.
Risk
management and strategy
With
the assistance of our IT vendor, we established policies and processes for assessing, identifying, and managing material risk
from cybersecurity threats and have integrated these processes into our overall risk management systems and processes. We routinely
assess material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our
information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems
or any information residing therein. Primary responsibility for assessing, monitoring and managing our cybersecurity risks rests
with an information technology (IT) consultant who reports to our Chief Financial Officer, to manage the risk assessment and
mitigation process. Our Board of Directors provides oversight to our cybersecurity efforts to ensure effective governance in
managing risks associated with cybersecurity threats. Our CFO provides periodic updates to the Board of Directors regarding our
cybersecurity program, including information about cyber risk management governance and status updates on various projects intended
to enhance the overall cybersecurity posture of the Company.
We
conduct periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our
business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include
identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such
risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks.
Following these risk assessments, responses may include re-design,
implementation, and maintenance of reasonable safeguards to minimize identified risks and address any identified gaps in existing safeguards;
and regularly monitoring of the effectiveness of our safeguards.
As
part of our overall risk management system, we monitor and test our safeguards and train our employees on these safeguards, in collaboration
with IT and management. Personnel at all levels and departments are made aware of our cybersecurity policies through trainings.
We
engage consultants, or other third parties in connection with our risk assessment processes. These service providers assist us to design
and implement our cybersecurity policies and procedures, as well as to monitor and test our safeguards.
We
have not encountered cybersecurity challenges that have materially impaired our operations or financial standing. For additional
information regarding risks from cybersecurity threats, please refer to “Cyber-attacks or other failures in
telecommunications or information technology systems and deficiency in our, or those of third parties upon which we rely,
cybersecurity could result in information theft, data corruption and significant disruption of our business operations.” under
Item 1A, “Risk Factors,” in this annual report on Form 10-K.
Governance
One
of the key functions of our board of directors is informed oversight of our risk management process, including risks from cybersecurity
threats. Our board of directors is responsible for monitoring and assessing strategic risk exposure, and our executive officers are responsible
for the day-to-day management of the material risks we face. Our board of directors administers its cybersecurity risk oversight function
directly as a whole, as well as through the audit committee.
Our
Chief Financial Officer is primarily responsible to assess and manage our material risks from cybersecurity threats with assistance from
third-party service providers.
Our
Chief Financial Officer oversees our cybersecurity policies and processes, including those described in “Risk Management and Strategy”
above. The cybersecurity risk management program includes tools and activities to prevent, detect, and analyze current and emerging cybersecurity
threats, and plans and strategies to address threats and incidents.
Item
2. Properties.
On
November 1, 2013 we entered into a sublease of approximately 17,000 square feet of mixed-use space of the facility located at 84 October
Hill Road, Suite 11, Holliston, Massachusetts, which is our corporate headquarters, from Harvard Bioscience. Our principal facilities
incorporate manufacturing, laboratory, development, sales and marketing, and administration functions. We believe our current facilities
are adequate for our needs for the foreseeable future.
Item
3. Legal Proceedings.
From
time to time, we may be involved in various claims and legal proceedings arising in the ordinary course of business. There are no
such matters pending that we expect to be material in relation to our business, financial condition, and results of operations
or cash flows.
Item
4. Mine Safety Disclosures.
Not
Applicable.
PART
II
Item
5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market
Information
The
Company filed an amendment (the “Certificate of Amendment”) to its Amended and Restated Certificate of Incorporation (the
“Certificate of Incorporation”) with the Secretary of State for the State of Delaware to change its name from Biostage, Inc.
to Harvard Apparatus Regenerative Technology, Inc. The Company also amended and restated its Amended and Restated Bylaws, solely to reflect
the name change (as amended, the “Third Amended and Restated Bylaws”). The Certificate of Amendment and the Third Amended
and Restated Bylaws each became effective on July 20, 2023.
In
connection with the name change, the Company traded on the OTCQB under the new ticker symbol “HRGN”. The new ticker symbol
was effective at the open of the market on July 20, 2023. Prior to that time, our common stock traded on the OTCQB under the symbol “BSTG.”
There
were 140 holders of record of our common stock as of March 18, 2024, which does not include persons or entities that hold their stock
in nominee or “street” name through various brokerage firms. We believe that the number of beneficial owners of our common
stock at that date was substantially greater.
Dividend
Policy
We
have never declared or paid cash dividends on our common stock in the past and do not intend to pay cash dividends on our common stock
in the foreseeable future. Any future determination to pay cash dividends will be at the discretion of our Board of Directors and will
depend on our financial condition, results of operations, capital requirements and other factors our Board of Directors deems relevant.
Recent
Sales of Unregistered Securities
During
the fiscal year ended December 31, 2023, all of our unregistered sales were previously disclosed in our Quarterly Reports on Form 10-Q
or in Current Reports on Form 8-K in relation to the applicable periods, which such issuances were done without registration under the
Securities Act in reliance on the exemptions provided by Section 4(a)(2) of the Securities Act as transactions not involving a public
offering and Rule 506 promulgated under the Securities Act as sales to an accredited investor, and in reliance on similar exemptions
under applicable state laws.
Item
6. Selected Financial Data.
Not
Applicable.
Item
7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Forward-Looking
Statements
The
following section of this Annual Report on Form 10-K entitled “Management’s Discussion and Analysis of Financial Condition
and Results of Operations” contains statements that are not statements of historical fact and are forward-looking statements within
the meaning of federal securities laws. These statements involve known and unknown risks, uncertainties and other factors that may cause
our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed
or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based
on assumptions and subject to risks and uncertainties.
In
some cases, you can identify forward-looking statements by terms such as “believe,” “may,” “estimate,”
“continue,” “anticipate,” “intend,” “should,” “could,” “would,”
“target,” “seek,” “aim,” “believe,” “predicts,” “think,” “objectives,”
“optimistic,” “new,” “goal,” “strategy,” “potential,” “is likely,”
“will,” “expect,” “plan” “project,” “permit” and similar expressions intended
to identify forward-looking statements. These statements reflect our current views with respect to future events, are based on assumptions
and are subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
We discuss many of these risks in greater detail in Item 1A.”Risk Factors” of this Annual Report on Form 10-K. You should
carefully review all of these factors, as well as the comprehensive discussion of forward-looking statements on page ii of this Annual
Report on Form 10-K.
Overview
We
are a clinical-stage biotechnology company focused on the development of regenerative medicine treatments for disorders of the gastro-intestinal
system and other organs that result from cancer, trauma or birth defects.
We
believe that our technology represents a next generation solution for restoring organ function because it allows the patient to regenerate
their own organ, thus eliminating the need for human donor or animal transplants, the sacrificing of another of the patient’s own
organs or permanent artificial implants.
Our
first esophageal product candidate, our esophageal implant was used in the first successful regeneration of the esophagus in a patient
with esophageal cancer. This successful first-in-human experience, plus the research we have performed on over 50 pigs, led the FDA to
approve our 10-patient phase 1 clinical trial. This combination trial will measure both safety and efficacy in the patient population.
We have contracted with IQVIA, a leading global provider
of advanced analytics, technology solutions and clinical research services to the life sciences industry, as the contract research organization
(CRO) to manage our first clinical trial. We activated the first clinical trial site and started screening patients in the third quarter
of 2023. Our product candidates are currently in development and have not yet received regulatory approval for sale anywhere in the world.
In addition to our development of regenerative medicine treatments, we
also sell longevity dietary supplements. In the second quarter of 2023, the Company’s subsidiary in Hong Kong, Harvard Apparatus
Regenerative Technology Limited, or Longevity Products, started focusing on longevity products. Longevity Products plans to include
a broad range of products focused on personal healthcare including longevity dietary supplements. Longevity Products started selling longevity supplements
in the third quarter of 2023. These products are marketed to the general public and initially targeted at consumers in the Great China
Region through eCommerce (online sales).
We
were incorporated and commenced operations on November 1, 2013 as a result of a spin-off from Harvard Bioscience, Inc., or Harvard Bioscience.
On that date, we became an independent company that operates the regenerative medicine business previously owned by Harvard Bioscience.
The spin-off was completed through the distribution of all the shares of common stock of Harvard Apparatus Regenerative Technology to
Harvard Bioscience stockholders.
We
continue to assess the market and regulatory approval pathway in China as to our implant products. We are not certain at this time as
to which market, including U.S. or China for example, may provide the most viable initial pathway for regulatory approval to a commercial
product. This will depend on a number of factors, including the approval and development processes, related costs, ability to raise capital
and the terms and conditions thereof, among other factors. Any development and capital raising efforts in China may include a joint venture
in relation to our Hong Kong subsidiary, and would also involve a number of commercial variables, including rights and obligations pertaining
to licensing, development, and financing, among others. Our failure to receive or obtain such clearances or approvals on a timely basis
or at all, whether that be in the U.S., China or otherwise, would have an adverse effect on our results of operations.
Since
our incorporation, we have devoted substantially all of our resources to developing our programs, building our intellectual property
portfolio, business planning, raising capital and providing selling, general and administrative support for these operations. To date,
we have financed our operations with proceeds from the sales of common stock and preferred stock. In December 2017, we sold the inventory
and rights to manufacture and sell research-only versions of our bioreactors to Harvard Bioscience.
We
have incurred substantial operating losses since our inception, and as of December 31, 2023 had an accumulated deficit of
approximately $92.0 million and will require additional financing to fund future operations. We expect that our operating cash
on-hand as of December 31, 2023 of approximately $0.4 million and debt financing of $0.5 million received in gross proceeds
subsequent to December 31, 2023 will enable us to fund our operating expenses and capital expenditure requirements into the second
quarter of 2024. We expect to continue to incur operating losses and negative cash flows from operations in future years. Therefore,
as disclosed in Note 1 to our Consolidated Financial Statements, these conditions raise substantial doubt about our ability to
continue as a going concern.
We
will need to raise additional funds to fund our operations. In the event we do not raise additional capital from outside sources
during the first quarter of 2024, we may be forced to curtail or cease our operations. Cash requirements and cash resource needs
will vary significantly depending upon the timing of the financial and other resource needs that will be required to complete
ongoing development, pre-clinical and clinical testing of product candidates, as well as regulatory efforts and collaborative
arrangements necessary for our products that are currently under development. We are currently seeking and will continue to seek
financings from other existing and/or new investors to raise necessary funds through a combination of public or private equity
offerings. We may also pursue debt financings, other financing mechanisms, research grants, or strategic collaborations and
licensing arrangements. We may not be able to obtain additional financing on favorable terms, if at all.
Our
operations will be adversely affected if we are unable to raise or obtain needed funding and may materially affect our ability to continue
as a going concern. Our consolidated financial statements have been prepared assuming that we will continue as a going concern and therefore,
the consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and
classification of assets or the amount and classifications of liabilities that may result from the outcome of this uncertainty.
Business Segments
The Company has two separate
reportable segments. The Company has one segment, Harvard Apparatus Regenerative Technology, Inc., or Regenerative Biotech, focused
on the development and commercialization of therapies to cure patients of cancers, injuries, and birth defects of the
gastro-intestinal tract and the airways. The other segment, Longevity Products, is focused on personal healthcare including
longevity dietary supplements.
2023
Financing Activities
During
the year ended December 31, 2023, we completed the following financing activities:
| |
● |
On
April 12, 2023 and on March 31, 2023, the Company entered into Securities Purchase Agreements, each a Purchase Agreement, with new
and existing investors, the Investors, pursuant to which the Investors purchased in a private placement an aggregate of
1,000,967 shares of common stock for the aggregate purchase price of approximately $6 million with a purchase price per unit of $6.00. |
2022
Financing Activities
During
the year ended December 31, 2022, we completed the following financing activities:
| |
● |
In
May 2022, we sold 854,771 shares of common stock and warrants to purchase 427,390 shares of common stock for the aggregate purchase
price of approximately $5.1 million and a purchase price per unit of $5.92. Each unit consisted of one share of common stock and
a warrant to purchase one half of one share of common stock, subject to adjustment as provided in the warrants. The warrants have
an exercise price of $8.88 per share, subject to adjustments as provided under the terms thereof, and were immediately exercisable.
The warrants are exercisable until five years (5) from the warrant issuance date. In May 2022, we also issued options to acquire
38,564 shares of common stock to satisfy sales commissions in the approximate amount of $155,660 incurred in relation to this private
placement. |
| |
● |
In
June 2022, the Company issued 4,000 shares of Series E Convertible Preferred Stock at a price of $1,000 per share to satisfy certain
indemnification obligations in the amount of $4.0 million, in lieu of paying cash. The Company issued an aggregate of 180 shares
of Series E Convertible Preferred Stock relating to accrued dividends during the year ended December 31, 2023. |
Management
Effective
as of November 26, 2021, we appointed David Green as Chief Executive Officer. Effective as of March 1, 2023, we transitioned the role
of Chief Executive Officer to Junli (Jerry) He, our existing director, and Mr. Green remains on our Board of Directors.
On
August 8, 2022, we appointed Joseph Damasio Jr. as Chief Financial Officer. In such role, Mr. Damasio serves as the Company’s principal
accounting officer and principal financial officer.
As of December 31, 2023, our consolidated business employed 18 individuals.
Components
of Operating Loss
Product
revenue. Product revenue consists of longevity product sales, launched in the Asia region in the third quarter of 2023. We had not
generated any revenue prior to the launch of our longevity products.
Research
and development expense. Research and development expense consists of salaries and related expenses, including share-based compensation,
for personnel and contracted consultants and various materials and other costs to develop our new products, primarily: synthetic scaffolds,
including investigation and development of materials and investigation and optimization of cellularization, as well as studies of cells
and cell behavior. Other research and development expenses include the costs of outside service providers and material costs for prototype
and test units and outside laboratories and testing facilities performing cell growth and materials experiments, as well as the costs
of all other preclinical research and testing including animal studies and expenses related to potential patents. We expense research
and development costs as incurred.
Sales
and marketing expense. Sales and marketing costs include advertising and payroll and related expenses for personnel engaged in marketing
and selling activities.
General
and administrative expense. General and administrative expense consists primarily of salaries and other related expenses,
including share-based compensation. Other costs include professional fees for legal and accounting services, insurance, investor relations
and facility costs.
Changes
in Fair Value of Warrant Liability. Changes in fair value of warrant liability represent the change in the fair value of outstanding
common stock warrants that were classified as liability awards during the year ended December 31, 2022. We used the Black-Scholes pricing
model to value the related warrant liability.
Critical
Accounting Estimates
Management’s
discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which
have been prepared in accordance with Generally Accepted Accounting Principles in the United States (U.S. GAAP). The preparation of these
consolidated financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenues,
expenses and related disclosures. We believe the following policies to be critical to the judgments and estimates used in the preparation
of our financial statements.
Share-based
Compensation
We
account for our share-based compensation in accordance with the fair value recognition provisions of current authoritative guidance.
Share-based awards, including stock options, are measured at fair value as of the grant date and recognized as expense over the requisite
vesting period (generally the service period), which we recognize on a straight-line basis. Expense on share-based awards for which vesting
is performance or milestone based is recognized on a straight-line basis from the date when we determine the achievement of the milestone
is probable to the vesting/milestone achievement date. We estimate the fair value of options granted using the Black-Scholes option valuation
model. Significant judgment is required in determining the proper assumptions used in these models. The assumptions used include the
risk-free interest rate, expected term, expected volatility and expected dividend yield. We base our assumptions on historical data when
available or, when not available, on a peer group of companies. However, these assumptions consist of estimates of future market conditions,
which are inherently uncertain and subject to our judgment, and therefore any changes in assumptions could significantly impact the future
grant date fair value of share-based awards.
Total
share-based compensation expense for each of the years ended December 31, 2023 and 2022 was approximately $3.5 million and $1.0 million,
respectively. Share-based compensation is further described in Note 15 to our Consolidated Financial Statements included in Item 15 of
this Annual Report on Form 10-K.
Warrant
Liability
Most
of the warrants to purchase shares of our common stock have been classified on our consolidated balance sheets as equity. We classify
warrants as a liability in our consolidated balance sheets if the warrant is a free-standing financial instrument that may require us
to transfer cash consideration upon exercise and that cash transfer event would be out of our control. Such a “liability warrant”
is initially recorded at fair value on the date of grant using the Black-Scholes model, net of issuance costs, and it is subsequently
re-measured to fair value at each subsequent balance sheet date. Changes in fair value of the warrant are recognized as a component of
other income (expense) in the consolidated statements of operations. We continued to adjust the liability for changes in fair value until
the expiration of the warrant liability in February 2022.
Results
of Operations
The
following table summarizes the results of our operations for the years ended December 31, 2023 and 2022 ($ in thousands):
| | |
Year Ended December 31, | | |
Change 2023 vs. 2022 | |
| | |
2023 | | |
2022 | | |
$ Change | | |
% | |
| Product revenue | |
$ | 103 | | |
$ | — | | |
$ | 103 | | |
| 100 | % |
| | |
| | | |
| | | |
| | | |
| | |
| Operating expenses | |
| | | |
| | | |
| | | |
| | |
| Cost of sales | |
| 24 | | |
| — | | |
| 24 | | |
| 100 | % |
| Research and development | |
| 3,062 | | |
| 1,742 | | |
| 1,320 | | |
| 76 | % |
| Sales and marketing | |
| 294 | | |
| — | | |
| 294 | | |
| 100 | % |
| General and administrative | |
| 5,713 | | |
| 4,411 | | |
| 1,302 | | |
| 30 | % |
| Total operating expenses | |
| 9,093 | | |
| 6,153 | | |
| 2,940 | | |
| 48 | % |
| | |
| | | |
| | | |
| | | |
| | |
| Other income (expense), net | |
| | | |
| | | |
| | | |
| | |
| Sublease income | |
| — | | |
| 87 | | |
| (87 | ) | |
| (100 | )% |
| Change in fair value of warrant liability | |
| — | | |
| 2 | | |
| (2 | ) | |
| (100 | )% |
| Interest income | |
| 64 | | |
| — | | |
| 64 | | |
| 100 | % |
| Interest expense | |
| (14 | ) | |
| (9 | ) | |
| (5 | ) | |
| (56 | )% |
| Other expense | |
| (5 | ) | |
| — | | |
| (5 | ) | |
| (100 | )% |
| Total other income, net | |
| 45 | | |
| 80 | | |
| (35 | ) | |
| (44 | )% |
| Net loss | |
$ | (8,945 | ) | |
$ | (6,073 | ) | |
$ | 2,872 | | |
| 47 | % |
Year
Ended December 31, 2023 Compared to Year Ended December 31, 2022
Product
Revenue
Product
revenue was $103,000 and zero for the year ended December 31, 2023 and 2022, respectively. Product revenue consists of longevity product
sales launched in the Asia region in the third quarter of 2023. We had not generated any revenue prior to the launch of our longevity
products.
Cost
of Sales
Cost
of sales was $24,000 and zero for the year ended December 31, 2023 and 2022, respectively. Cost of sales consists of the purchase price
of consumer products, taxes, inbound and outbound shipping costs.
Research
and Development Expense
Research
and development expense increased approximately $1.3 million, or 76%, to approximately $3.1 million for the year ended December 31, 2023
as compared to approximately $1.7 million for the year ended December 31, 2022. This was due primarily to higher headcount and preclinical trial activities to increase our product pipeline and clinical trial
activities resulting in our first site activation in the third quarter of 2023.
Sales
and Marketing Expense
Longevity
Products launched its longevity products business in the second quarter of 2023 so there were no prior period costs. Selling and
marketing expense was $0.3 million for the year ended December 31, 2023 as compared to zero for the comparable period.
General
and Administrative Expense
General
and administrative expense increased approximately $1.3 million, or 30%, to approximately $5.7 million for the year ended December
31, 2023 as compared to approximately $4.4 million for the year ended December 31, 2022. This increase was primarily due to
share-based compensation expense of $2.4 million and increased headcount related costs of approximately $0.6 million offset by a
decrease of approximately $0.4 million for supporting our ongoing public company requirements and reduced legal and related costs of
approximately $1.3 million relating to the completion of litigation for a wrongful death complaint and related matters more fully
described in Note 9 to our consolidated financial statements.
Sublease
Income
On
January 5, 2022, the Company executed a four-month sublease agreement for certain laboratory and office space at its Holliston, Massachusetts
facility. The Company further extended the sublease agreement to a month-to-month basis until August 31, 2022, when the other party vacated
the premises. For the year ended December 31, 2022, the Company recorded sublease income of approximately $87,000 relating to this agreement.
We had no sublease agreements generating sublease income for the year ended December 31, 2023.
Change
in Fair Value of Warrant Liability
For
the year ended December 31, 2022, the change in fair value of our warrant liability resulted in other income of approximately $2,000.
These warrants expired unexercised in February of 2022.
Interest
income
During
the year ended December 31, 2023, we recorded interest income of approximately $64,000 earned from our money market account and certificate
of deposit. During the year ended December 31, 2022, we received minimal interest income from cash accounts.
Interest expense
During the year ended December 31, 2023, we recorded
interest expense of approximately $14,000 on insurance installment payments. During the year ended December 31, 2022, we recorded interest
expense of approximately $9,000 on insurance installment payments.
Liquidity
and Capital Resources
Sources
of Liquidity. We have incurred operating losses since inception and as of December 31, 2023, we had an accumulated deficit of approximately
$92.0 million. We are currently investing significant resources in the development and commercialization of our product candidates for
use by clinicians and researchers in the field of regenerative medicine. As a result, we expect to incur operating losses and negative
operating cash flows for the foreseeable future.
Operating
Activities. Net cash used in operating activities of approximately $6.9 million for the year ended December 31, 2023 was due
primarily to our net loss of approximately $8.9 million offset by adjustments for non-cash items of approximately $3.6 million due
to non-cash expenses for share-based compensation, depreciation and amortization, and an approximately $1.6 million decrease to cash from changes in
working capital due to the timing of payments for accounts receivable, inventory, prepaid expenses, deferred financing costs,
long-term prepaid contracts, accounts payable and accrued expenses.
Net
cash used in operating activities of approximately $5.1 million for the year ended December 31, 2022 was primarily a result of our
net loss of approximately $6.1 million and $0.6 million for deferred financing costs, offset by approximately $1.1 million of
non-cash items related to share-based compensation, depreciation and amortization, and an increase of approximately $0.5 million of cash provided
from working capital due to the timing of prepaid expenses, accounts payable, and accrued and other current liabilities.
Investing
Activities. Net cash used in investing activities for the years ended December 31, 2023 and 2022 totaled $11,000 and $5,000, respectively,
and represented purchases of property, plant and equipment. During the year ended December 31, 2023, we invested in a certificate of
deposit for $2.5 million. We withdrew $1.3 million from the certificate of deposit prior to the maturity date to pay clinical trial related
deposits. The certificate of deposit matured in October 2023 with the remaining $1.2 million released from short-term investments into
cash and cash equivalents.
Financing
Activities. Net cash generated from financing activities was approximately $6.1 million during the year ended December 31, 2023 and
consisted of net proceeds received from a private placement transaction for the issuance of common stock and stock option exercises. Net cash generated from financing activities was approximately $5.1 million during the year ended December
31, 2022 and consisted of net proceeds received from private placement transactions for the issuance of common stock and warrants to
purchase common stock.
We
continue to pursue our esophageal program, including advancing to operate as a clinical stage company. Given our current limited cash
resources, we intend to closely monitor our cash expenses as such cash resources are expected to only allow us to meet our operating
needs into the second quarter of 2024.
We have incurred substantial operating losses since
our inception, and as of December 31, 2023 had an accumulated deficit of approximately $92.0 million and will require additional financing
to fund future operations. We expect that our operating cash on-hand as of December 31, 2023 of approximately $0.4 million and debt financing
of $0.5 million received in gross proceeds subsequent to December 31, 2023 will enable us to fund our operating expenses and capital expenditure
requirements into the second quarter of 2024. We expect to continue to incur operating losses and negative cash flows from operations
in future years. Therefore, as disclosed in Note 1 to our Consolidated Financial Statements, these conditions raise substantial doubt
about our ability to continue as a going concern.
Recently
Issued Accounting Pronouncements
A
description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations
is disclosed in Note 2 to our consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K.
Off-Balance
Sheet Arrangements
We
did not have, during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under applicable
Securities and Exchange Commission rules.
Item
7A. Quantitative and Qualitative Disclosures about Market Risk.
Not
Applicable.
Item
8. Financial Statements and Supplementary Data.
The
information required by this item is contained in the consolidated financial statements filed as part of this Annual Report on Form 10-K
listed under Item 15 of Part IV below.
Item
9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item
9A. Controls and Procedures.
This
Annual Report on Form 10-K includes the certifications of our principal executive officer and principal financial officer required by
Rule 13a-14 of the Securities Exchange Act of 1934, as amended, or the Exchange Act. See Exhibits 31.1 and 31.2. This Item 9A includes
information concerning the controls and control evaluations referred to in those certifications.
(a)
Evaluation of Disclosure Controls and Procedures
Disclosure
controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) are designed to ensure that information
required to be disclosed in reports filed or submitted under the Exchange Act is recorded, processed, summarized, and reported within
the time periods specified in Securities and Exchange Commission’s rules and forms and that such information is accumulated and
communicated to management, including our principal executive officer and principal financial officer, to allow timely decisions regarding
required disclosures. Based on the evaluation, our principal executive and principal financial officers concluded that, as of December
31, 2023, our disclosure controls and procedures were effective.
In
connection with the preparation of this Annual Report on Form 10-K, our management, under the supervision and with the participation
of our principal executive officer and principal financial officer, conducted an evaluation of the effectiveness of the design and operation
of our disclosure controls and procedures as of December 31, 2023. Our disclosure controls and procedures are designed to provide reasonable
assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed,
summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and our
management necessarily was required to apply its judgment in evaluating and implementing our disclosure controls and procedures. Based
upon the evaluation described above, our principal executive officer and principal financial officer have concluded that they believe
that our disclosure controls and procedures were effective, as of the end of the period covered by this report, in providing reasonable
assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated
and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions
regarding required disclosures, and is recorded, processed, summarized and reported within the time periods specified in the Securities
and Exchange Commission’s rules and forms.
(b)
Management’s Annual Report on Internal Control Over Financial Reporting
Our
management, under the supervision of the principal executive officer and the principal financial officer, is responsible for establishing
and maintaining an adequate system of internal control over financial reporting. Internal control over financial reporting (as defined
in Rules 13a-15(f) and 15d(f) under the Exchange Act) is a process designed to provide reasonable assurance regarding the reliability
of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP.
A
company’s internal control over financial reporting includes those policies and procedures that: (a) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (b) provide reasonable
assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with U.S.
GAAP; (c) provide reasonable assurance that receipts and expenditures are being made only in accordance with appropriate authorization
of management and the Board of Directors; and (d) provide reasonable assurance regarding prevention or timely detection of unauthorized
acquisition, use, or disposition of our assets that could have a material effect on the consolidated financial statements.
Due
to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of
any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions,
or that the degree of compliance with the policies or procedures may deteriorate.
In
connection with the preparation of this Annual Report on Form 10-K, our management conducted an evaluation of the effectiveness of our
internal control over financial reporting as of December 31, 2023 based on the criteria established in Internal Control - Integrated
Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO. As a result of that evaluation,
management has concluded that our internal control over financial reporting was effective as of December 31, 2023.
As
a smaller reporting company, we are exempt from the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002.
As a result, Marcum LLP, our independent registered public accounting firm, has not audited or issued an attestation report with respect
to the effectiveness of our internal control over financial reporting as of December 31, 2023.
(c)
Changes in Internal Controls Over Financial Reporting
Our
management, with the participation of the principal executive officer and the principal financial officer, has evaluated whether any
change in our internal control over financial reporting occurred during the fourth quarter ended December 31, 2023. Except as noted above,
management concluded that there were no changes in our internal controls over financial reporting during the quarter ended December 31,
2023 that materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
(d)
Inherent Limitations on Effectiveness of Controls
The
design of any system of control is based upon certain assumptions about the likelihood of future events, and there can be no assurance
that any design will succeed in achieving its stated objectives under all future events, no matter how remote, that controls may become
inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may not deteriorate. Because
of their inherent limitations, systems of control may not prevent or detect all misstatements. Accordingly, even effective systems of
control can provide only reasonable assurance of achieving their control objectives.
Item
9B. Other Information.
None.
PART
III
Item
10. Directors, Executive Officers and Corporate Governance.
Incorporated
by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our
2024 Annual Meeting of Stockholders. Information concerning executive officers of our company is included in Part I of this Annual Report
on Form 10 K as Item 1. Business - Information about our Executive Officers and incorporated herein by reference.
Item
11. Executive Compensation.
Incorporated
by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our
2024 Annual Meeting of Stockholders.
Item
12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Incorporated
by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our
2024 Annual Meeting of Stockholders.
Item
13. Certain Relationships and Related Transactions, and Director Independence.
Incorporated
by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our
2024 Annual Meeting of Stockholders.
Item
14. Principal Accounting Fees and Services.
Our
independent public accounting firm is Marcum LLP, Boston, Massachusetts, PCAOB Auditor ID 688.
Incorporated
by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our
2024 Annual Meeting of Stockholders.
PART
IV
Item
15. Exhibits, Financial Statement Schedules.
| (a) |
Documents
Filed. The following documents are filed as part of this Annual Report on Form 10-K: |
(1)
Financial Statements. The consolidated financial statements of Harvard Apparatus Regenerative Technology, Inc. and its subsidiaries filed
under this Item 15:
(2)
Financial Statement Schedules: None. Financial statement schedules have been omitted since the required information is included in our
consolidated financial statements contained elsewhere in this Annual Report on Form 10-K.
(3)
Exhibits. The exhibits listed in the accompanying Exhibit Index are filed as a part of this Annual Report on Form 10-K.
| (b) |
Exhibits:
The exhibits listed in the accompanying Exhibit Index are filed as a part of this Annual Report on Form 10-K. |
| |
|
| (c) |
Separate
Financial Statements and Schedules: None. Financial statement schedules have been omitted since the required information is included
in our consolidated financial statements contained elsewhere in this Annual Report on Form 10-K. |
INDEX
TO CONSOLIDATED FINANCIAL STATEMENTS
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC.
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Shareholders and Board of Directors of
Harvard
Apparatus Regenerative Technology, Inc. and Subsidiaries
Opinion
on the Financial Statements
We
have audited the accompanying consolidated balance sheets of Harvard Apparatus Regenerative Technology, Inc. and Subsidiaries (the
“Company”) (formerly known as Biostage, Inc.) as of December 31, 2023 and 2022, the related consolidated statements of
operations, changes in stockholders’ equity (deficit) and cash flows for each of the two years in the period ended December 31, 2023, and the related notes
(collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in
all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and
its cash flows for each of the two years in the period ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of
America.
Explanatory
Paragraph – Going Concern
The
accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more
fully described in Note 1, the Company has suffered recurring losses from operations, has an accumulated deficit, uses cash flows in
its operations, and will require additional financing to continue to fund its operations. These conditions raise substantial doubt about
the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in
Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis
for Opinion
These
financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s
financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board
(United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal
securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We
conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain
reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company
is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits
we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our
audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error
or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence
regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles
used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We
believe that our audits provide a reasonable basis for our opinion.
Critical
Audit Matter
The
critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated
or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial
statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter
does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit
matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Share-Based
Compensation – Performance-Based Awards
Description
of the Matter
As
described in Note 14 to the consolidated financial statements, the Company has 773,195 unvested performance-based options outstanding
for which there is unrecognized compensation expense of approximately $2.8 million at December 31, 2023. No expense has been recognized
for these unvested awards as of December 31, 2023 given that the milestone achievements for these awards have not yet been deemed probable
for accounting purposes. As described in Note 2 to the consolidated financial statements, the Company measures all stock options and
restricted stock awards granted to employees, directors and non-employees based on the fair value on the date of the grant and recognizes
compensation expense of those awards, net of estimated forfeitures, over the requisite vesting period. Expense on share-based awards
for which vesting is performance or milestone based is recognized on a straight-line basis from the date when it is determined that the
achievement of the milestone is probable to the vesting/milestone achievement date.
We
identified the Company’s expense recognition for share-based awards that contain performance-based vesting provisions as a critical
audit matter. The principal considerations for our determination that the expense recognition for share-based awards that contain performance-based
vesting provision awards is a critical audit matter are the assumptions and risk of bias related to the conclusion of the probability
of achievement of the performance conditions impacting vesting of the awards, or more specifically, the achievement of the business milestones,
as defined in the grant agreements. Auditing management’s assumptions regarding the probability of achievement of the business
milestones defined in the grant agreements was complex and required a high degree of auditor judgment and increased audit effort.
How
We Addressed the Matter in Our Audit
Our
audit procedures related to the expense recognition of share-based awards that contain performance-based vesting provisions included
the following, among others, (i) obtaining and analyzing the grant agreements for outstanding share-based awards with performance-based
vesting provisions, (ii) recalculated the total outstanding share-based awards with performance-based vesting provisions at year-end
based upon cumulative grants, net of cumulative forfeitures, and (iii) discussed with management and evaluated their conclusions reached
on the probability of achievement of the business milestones within the performance based awards by assessing the Company’s liquidity
requirements needed to fund the achievement of the milestones outlined in the grant agreements and reviewed the Company’s public
press releases through the issuance date of these financials.
Marcum
LLP
We
have served as the Company’s auditor since 2022.
Boston,
MA
March 28, 2024
(PCAOB
ID # 688)
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC. AND SUBSIDIARIES
CONSOLIDATED
BALANCE SHEETS
(In
thousands, except share and par value data)
| | |
2023 | | |
2022 | |
| | |
December 31, | | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
| | |
| |
| ASSETS | |
| | | |
| | |
| Current assets: | |
| | | |
| | |
| Cash and cash equivalents | |
$ | 432 | | |
$ | 1,241 | |
| Accounts receivable | |
| 4 | | |
| — | |
| Inventory | |
| 50 | | |
| — | |
| Prepaid research and development | |
| 210 | | |
| 274 | |
| Prepaid expenses and other current assets | |
| 87 | | |
| 79 | |
| Total current assets | |
| 783 | | |
| 1,594 | |
| Property, plant and equipment, net | |
| 25 | | |
| 49 | |
| Right-of-use assets, net | |
| 48 | | |
| 147 | |
| Deferred financing costs | |
| 544 | | |
| 610 | |
| Long-term prepaid contracts | |
| 1,214 | | |
| — | |
| Total assets | |
$ | 2,614 | | |
$ | 2,400 | |
| | |
| | | |
| | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 445 | | |
$ | 682 | |
| Accrued and other current liabilities | |
| 475 | | |
| 582 | |
| Operating lease liability, current | |
| 48 | | |
| 99 | |
| Total current liabilities | |
| 968 | | |
| 1,363 | |
| Operating lease liability, net of current portion | |
| — | | |
| 48 | |
| Total liabilities | |
| 968 | | |
| 1,411 | |
| | |
| | | |
| | |
| Commitments and contingencies (Note 9) | |
| - | | |
| | |
| Series E convertible preferred stock, par value $0.01 per share, 5,000 shares authorized; 0 and 4,180 shares issued and outstanding at December 31, 2023 and 2022, respectively | |
| — | | |
| 4,180 | |
| | |
| | | |
| | |
| Stockholders’ equity (deficit): | |
| | | |
| | |
| Common stock, par value $0.01 per share, 60,000,000 shares authorized; 13,947,324 and 12,174,467 issued and outstanding at December 31, 2023 and 2022, respectively | |
| 139 | | |
| 122 | |
| Additional paid-in capital | |
| 93,463 | | |
| 79,698 | |
| Accumulated deficit | |
| (91,956 | ) | |
| (83,011 | ) |
| Total stockholders’ equity (deficit) | |
| 1,646 | | |
| (3,191 | ) |
| Total liabilities and stockholders’ equity (deficit) | |
$ | 2,614 | | |
$ | 2,400 | |
See
accompanying notes to consolidated financial statements.
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF OPERATIONS
(In
thousands, except share and per share data)
| | |
2023 | | |
2022 | |
| | |
Year Ended | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| Product revenue | |
$ | 103 | | |
$ | — | |
| | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | |
| Cost of sales | |
| 24 | | |
| — | |
| Research and development | |
| 3,062 | | |
| 1,742 | |
| Sales and marketing | |
| 294 | | |
| — | |
| General and administrative | |
| 5,713 | | |
| 4,411 | |
| Total operating expenses | |
| 9,093 | | |
| 6,153 | |
| | |
| | | |
| | |
| Operating loss | |
| (8,990 | ) | |
| (6,153 | ) |
| | |
| | | |
| | |
| Other income (expense), net: | |
| | | |
| | |
| Sublease income | |
| — | | |
| 87 | |
| Change in fair value of warrant liability | |
| — | | |
| 2 | |
| Interest income | |
| 64 | | |
| — | |
| Interest expense | |
| (14 | ) | |
| (9 | ) |
| Other expense | |
| (5 | ) | |
| — | |
| Total other income, net | |
| 45 | | |
| 80 | |
| | |
| | | |
| | |
| Net loss | |
| (8,945 | ) | |
| (6,073 | ) |
| Preferred stock dividends | |
| (77 | ) | |
| (180 | ) |
| Net loss attributable to common stockholders | |
$ | (9,022 | ) | |
$ | (6,253 | ) |
| | |
| | | |
| | |
| Basic and diluted net loss per share attributable to common stockholders | |
$ | (0.67 | ) | |
$ | (0.55 | ) |
| Weighted average common shares outstanding, basic and diluted | |
| 13,455,666 | | |
| 11,349,610 | |
See
accompanying notes to consolidated financial statements.
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIT)
(In
thousands, except share data)
| | |
Series E Convertible Preferred Stock | | |
Number of Common Shares Outstanding | | |
Common Stock | | |
Additional Paid-in Capital | | |
Accumulated Deficit | | |
Total Stockholders Equity (Deficit) | |
| | |
Series E Convertible Preferred Stock | | |
Number of Common Shares Outstanding | | |
Common Stock | | |
Additional Paid-in Capital | | |
Accumulated Deficit | | |
Total Stockholders’ Equity (Deficit) | |
| Balance at January 1, 2022 | |
$ | — | | |
| 10,760,871 | | |
$ | 108 | | |
$ | 73,801 | | |
$ | (76,938 | ) | |
$ | (3,029 | ) |
| Net loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| (6,073 | ) | |
| (6,073 | ) |
| Share-based compensation | |
| — | | |
| — | | |
| — | | |
| 1,031 | | |
| — | | |
| 1,031 | |
| Issuance of series E convertible preferred stock | |
| 4,000 | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| Preferred stock dividends | |
| 180 | | |
| — | | |
| — | | |
| (180 | ) | |
| — | | |
| (180 | ) |
| Issuance of common stock and warrants to purchase common stock | |
| — | | |
| 854,771 | | |
| 8 | | |
| 5,052 | | |
| — | | |
| 5,060 | |
| Issuance of common stock from exercise of warrants | |
| — | | |
| 558,825 | | |
| 6 | | |
| (6 | ) | |
| — | | |
| — | |
| Balance at December 31, 2022 | |
| 4,180 | | |
| 12,174,467 | | |
| 122 | | |
| 79,698 | | |
| (83,011 | ) | |
| (3,191 | ) |
| Net loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| (8,945 | ) | |
| (8,945 | ) |
| Share-based compensation | |
| — | | |
| — | | |
| — | | |
| 3,461 | | |
| — | | |
| 3,461 | |
| Conversion of preferred stock for common stock | |
| (4,257 | ) | |
| 706,626 | | |
| 7 | | |
| 4,250 | | |
| — | | |
| 4,257 | |
| Preferred stock dividends | |
| 77 | | |
| — | | |
| — | | |
| (77 | ) | |
| — | | |
| (77 | ) |
| Issuance of common stock | |
| — | | |
| 1,000,967 | | |
| 10 | | |
| 5,982 | | |
| — | | |
| 5,992 | |
| Issuance of common stock from exercise of options | |
| — | | |
| 65,264 | | |
| — | | |
| 149 | | |
| — | | |
| 149 | |
| Balance at December 31, 2023 | |
$ | — | | |
| 13,947,324 | | |
$ | 139 | | |
$ | 93,463 | | |
$ | (91,956 | ) | |
$ | 1,646 | |
See
accompanying notes to consolidated financial statements.
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF CASH FLOWS
(In
thousands)
| | |
2023 | | |
2022 | |
| | |
Year ended December 31, | |
| | |
2023 | | |
2022 | |
| OPERATING ACTIVITIES | |
| | | |
| | |
| Net loss | |
$ | (8,945 | ) | |
$ | (6,073 | ) |
| Adjustments to reconcile net loss to net cash and cash equivalents used in operating activities: | |
| | | |
| | |
| Share-based compensation expense | |
| 3,461 | | |
| 1,031 | |
| Depreciation | |
| 35 | | |
| 52 | |
| Amortization of operating right-of-use assets | |
| 99 | | |
| 22 | |
| Change in fair value of warrant liability | |
| — | | |
| (2 | ) |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Accounts receivable | |
| (4 | ) | |
| — | |
| Inventory | |
| (50 | ) | |
| — | |
| Prepaid research and development | |
| 64 | | |
| (274 | ) |
| Prepaid expenses and other current assets | |
| (8 | ) | |
| 216 | |
| Deferred financing costs | |
| 66 | | |
| (610 | ) |
| Long-term prepaid contracts | |
| (1,214 | ) | |
| — | |
| Accounts payable | |
| (237 | ) | |
| 6 | |
| Operating lease liability | |
| (99 | ) | |
| (22 | ) |
| Accrued and other current liabilities | |
| (107 | ) | |
| 548 | |
| Net cash used in operating activities | |
| (6,939 | ) | |
| (5,106 | ) |
| | |
| | | |
| | |
| INVESTING ACTIVITIES | |
| | | |
| | |
| Purchases of short-term investments | |
| (2,523 | ) | |
| — | |
| Redemption of short-term investments | |
| 2,523 | | |
| — | |
| Purchases of property, plant and equipment | |
| (11 | ) | |
| (5 | ) |
| Net cash used in investing activities | |
| (11 | ) | |
| (5 | ) |
| | |
| | | |
| | |
| FINANCING ACTIVITIES | |
| | | |
| | |
| Proceeds from issuance of common stock | |
| 5,992 | | |
| 5,060 | |
| Proceeds from exercise of stock options | |
| 149 | | |
| — | |
| Net cash provided by financing activities | |
| 6,141 | | |
| 5,060 | |
| Net decrease in cash and cash equivalents | |
| (809 | ) | |
| (51 | ) |
| Cash and cash equivalents at the beginning of the year | |
| 1,241 | | |
| 1,292 | |
| Cash and cash equivalents at the end of the year | |
$ | 432 | | |
$ | 1,241 | |
| | |
| | | |
| | |
| SUPPLEMENTAL INFORMATION | |
| | | |
| | |
| Interest paid in cash | |
$ | 14 | | |
$ | 9 | |
| | |
| | | |
| | |
| Supplemental disclosure of non-cash activities: | |
| | | |
| | |
| Settlement of contingency matter | |
$ | — | | |
$ | (3,250 | ) |
| Settlement of due to Harvard Bioscience included in accrued and other current liabilities | |
$ | — | | |
$ | (750 | ) |
| Issuance of Series E convertible preferred stock | |
$ | — | | |
$ | 4,000 | |
| Preferred stock dividends | |
$ | 77 | | |
$ | 180 | |
| Increase of right-of-use asset and liability due to lease extension | |
$ | — | | |
$ | 63 | |
See
accompanying notes to consolidated financial statements.
HARVARD
APPARATUS REGENERATIVE TECHNOLOGY, INC. AND SUBSIDIARIES
NOTES
TO CONSOLIDATED FINANCIAL STATEMENTS
Years
Ended December 31, 2023 and 2022
1.
Organization
Overview
Harvard
Apparatus Regenerative Technology, Inc. (Harvard Apparatus Regenerative Technology or the Company) is a biotechnology company with a
mission to cure patients of cancers, injuries, and birth defects of the gastro-intestinal tract and the airways. The Company believes
its technology is likely to be used to treat esophageal cancer, esophageal injuries, and birth defects in the esophagus. The Company
believes additional product candidates in its pipeline may treat intestinal cancer and colon cancer. Since inception, the Company has
devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, and
acquiring operating assets.
On
October 31, 2013, Harvard Bioscience, Inc., or Harvard Bioscience, contributed its regenerative medicine business assets, plus $15 million
of cash, into Harvard Apparatus Regenerative Technology, or the Separation. On November 1, 2013, the spin-off of the Company from Harvard
Bioscience was completed. On that date, the Company became an independent company that operates the regenerative medicine business previously
owned by Harvard Bioscience. The spin-off was completed through the distribution to Harvard Bioscience stockholders of all the shares
of common stock of Harvard Apparatus Regenerative Technology, or the Distribution.
Basis
of Presentation
The
consolidated financial statements reflect the Company’s financial position, results of operations and cash flows in conformity
with generally accepted accounting principles in the United States, or U.S. GAAP.
Going
Concern
The
Company has incurred substantial operating losses since its inception, and as of December 31, 2023 had an accumulated deficit of
approximately $92.0
million and will require additional financing to fund future operations. The Company expects that its operating cash on-hand as of
December 31, 2023 of approximately $0.4
million and debt financing of $0.5
million in gross proceeds received subsequent to December 31, 2023 will enable it to fund its operating expenses and capital
expenditure requirements only into the second quarter of 2024. Therefore, these conditions raise substantial doubt about the
Company’s ability to continue as a going concern.
The
Company will need to raise additional funds to fund its operations. In the event the Company does not raise additional capital from outside
sources during the first quarter of 2024, it may be forced to curtail or cease its operations. Cash requirements and cash resource
needs will vary significantly depending upon the timing of the financial and other resource needs that will be required to complete ongoing
development, pre-clinical and clinical testing of product candidates, as well as regulatory efforts and collaborative arrangements necessary
for the Company’s product candidates that are currently under development. The Company is currently seeking and will continue to
seek financings from other existing and/or new investors to raise necessary funds through a combination of public or private equity offerings.
The Company may also pursue debt financings, other financing mechanisms, research grants, or strategic collaborations and licensing arrangements.
The Company may not be able to obtain additional financing on favorable terms, if at all.
The
Company’s operations will be adversely affected if it is unable to raise or obtain needed funding and may materially affect the
Company’s ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming
that the Company will continue as a going concern and therefore, the consolidated financial statements do not include any adjustments
to reflect the possible future effects on the recoverability and classification of assets or the amount and classifications of liabilities
that may result from the outcome of this uncertainty.
2.
Summary of Significant Accounting Policies
Principles
of Consolidation
The
consolidated financial statements include the accounts of Harvard Apparatus Regenerative Technology, Inc. (Regenerative Biotech) and its three
wholly-owned subsidiaries, Harvard Apparatus Regenerative Technology Limited (Hong Kong), Harvard Apparatus Regenerative Technology
(Hangzhou) Limited (China) and Harvard Apparatus Regenerative Technology GmbH (Germany). All intercompany balances and transactions
have been eliminated in consolidation.
Use
of Estimates
The
process of preparing consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Such estimates include, but are not
limited to, share-based compensation, valuation of warrant liability, accrued expenses and the valuation allowance for deferred income
taxes. Actual results could differ from those estimates.
Revenue
We
recognize revenue in accordance with FASB ASC 606, Revenue from Contracts with Customers. We offer consumer products primarily
through a third-party online store. Revenue is recognized at a point in time when control of the goods is transferred to the customer,
which generally occurs upon the delivery to the customer. For any company direct sales to customers, revenue is recognized at a point
in time upon shipment of product or hand-delivery to customer. Revenue also excludes any amounts collected on behalf of third parties,
including sales and indirect taxes.
We
identify a performance obligation as distinct if both the following criteria are true: the customer can benefit from the good or service
either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer
the good or service to the customer is separately identifiable from other promises in the contract. Determining the standalone selling
price (“SSP”) and allocation of consideration from a contract to the individual performance obligations, and the appropriate
timing of revenue recognition, is the result of significant qualitative and quantitative judgments. Management considers a variety of
factors such as historical sales, usage rates, costs, and expected margin, which may vary over time depending upon the unique facts and
circumstances related to each performance obligation in making these estimates. While changes in the allocation of the SSP between performance
obligations will not affect the amount of total revenue recognized for a particular contract, any material changes could impact the timing
of revenue recognition, which would have a material effect on our financial position and result of operations. This is because the contract
consideration is allocated to each performance obligation, delivered or undelivered, at the inception of the contract based on the SSP
of each distinct performance obligation.
Cost
of Sales
Cost
of sales primarily consists of the purchase price of consumer products, taxes, inbound and outbound shipping costs. Shipping costs to
receive products from our suppliers are recognized as cost of sales when incurred. E-commerce processing and related transaction costs,
including those associated with seller transactions, are classified in sales and marketing on our consolidated statements of
operations.
Research
and Development
Research
and development costs are expensed as incurred.
Sales
and Marketing
Sales
and marketing costs include advertising and payroll and related expenses for personnel engaged in marketing and selling activities.
General
and Administrative
General
and administrative expenses primarily consist of costs for corporate functions, including payroll and related expenses; facilities and
equipment expenses, such as depreciation and amortization expense and rent; and professional fees.
Segment Information
The
Company manages its operations as two separate operating segments for the purposes of assessing performance and making operating
decisions. The Company has one operating unit focused on the development and commercialization of therapies to cure patients of
cancers, injuries, and birth defects of the gastro-intestinal tract and the airways. The other operating unit is focused on personal
healthcare through longevity dietary supplements. We have determined that our chief executive officer is the chief operating
decision maker (CODM). The CODM reviews financial information presented by operating unit. Resource allocation decisions are
made by the CODM based on operating unit results.
Cash
and Cash Equivalents
The
Company considers all highly liquid investments with a maturity of three months or less at the date of purchase to be cash
equivalents. The Company currently invests available cash in money market funds. As of December 31, 2023, the Company had
approximately $111,000
of cash equivalents in a money market fund.
Accounts Receivable
Allowances
are provided for estimated amounts of accounts receivable which may not be collected. At
December 31, 2023,
we determined that no allowance against accounts receivable was necessary.
Inventory
Inventory,
consisting of products available for sale, are primarily accounted for using the first-in, first-out method, and are valued at the lower
of cost or net realizable value.
We
maintain ownership of our inventory at the third-party warehouse, regardless of whether fulfillment is provided by us or the third-party
e-commerce seller, and therefore these products are included in our inventories.
Deferred Financing Costs
We capitalized costs relating to a
registered offering that we postponed in 2023 but expect to resume in the near future. The costs include payments made to attorneys,
accountants, regulators and consultants. Once we complete the registered offering, the deferred financing costs will be reclassified
to stockholders’ equity (deficit) on the consolidated balance sheets to offset the proceeds from the registered offering.
Long-term
prepaid contracts
We
have contracted with partners relating to our clinical trial activities. Upon execution of the contracts, we made initial payments
of $1.2
million as deposits recorded as long-term assets and will be applied against final invoices which are more than a year away. The deposits will be recorded as expense when the clinical trial is substantially
completed. Costs for the clinical trial activities throughout our clinical trial under these contracts are recognized as expense and payable
based on costs incurred.
Property,
Plant and Equipment
Property,
plant and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets
as follows:
Schedule of Property Plant and Equipment Estimated Useful Lives
| Leasehold improvements | |
Shorter of expected useful life
or lease term |
| Computer equipment and software | |
3 years |
| Furniture, machinery and equipment | |
5-7 years |
Maintenance
and repairs are charged to expense as incurred, while any additions or improvements are capitalized.
Impairment
of Long-Lived Assets
Assessments
of long-lived assets and the remaining useful lives of such long-lived assets are reviewed for impairment whenever a triggering event
occurs or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. An asset, or group of assets,
are considered to be impaired when the undiscounted estimated net cash flows expected to be generated by the asset, or group of assets,
are less than its carrying amount. The impairment recognized is the amount by which the carrying amount exceeds the fair market value
of the impaired asset, or group of assets, based on the present value of the expected future cash flows associated with the use of the
asset. Through December 31, 2023, no such impairment charges have been recorded.
Share-based
Compensation
The
Company measures all stock options and restricted stock awards granted to employees, directors and non-employees based on the fair value
on the date of the grant and recognizes compensation expense of those awards, net of forfeitures, over the requisite vesting period,
which is generally the service period of the respective award. Generally, the Company issues stock options and restricted stock awards
with only service-based vesting conditions on a straight-line basis over the requisite service period for the entire award (that is,
over the requisite service period of the last separately vesting portion of the award). Expense on share-based awards for which vesting
is performance or milestone based is recognized on a straight-line basis from the date when it is determined that the achievement of
the milestone is probable to the vesting/milestone achievement date.
The
Company elected to use the Black-Scholes option-pricing model for the valuation of stock-based payment awards. The determination of the
fair value of stock-based payment awards is determined on the date of grant using the Black-Scholes option-pricing model which is affected
by the market price as well as assumptions regarding a number of subjective variables. These variables include, but are not limited to,
its expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. When
performance-based grants are issued, the Company recognizes no expense until achievement of the performance requirement is deemed probable.
Share-based
compensation expense is based on awards ultimately expected to vest and has been reduced for annualized estimated forfeiture where the
minimum amount of expense recorded is at least equal to the percent of an award vested. Forfeitures are estimated based on historical
experience and weighting of various employee classes under the respective plan at the time of grant and revised, if necessary, in subsequent
periods if actual forfeitures differ from those estimates. Until December 31, 2022, we estimated forfeitures at the time of grant
and would revise our estimate, if necessary, in subsequent periods. As of January 1, 2023, we account for forfeitures as they occur.
The
fair value of Restricted Stock Units, or RSUs, is based on the number of shares granted and market price of the stock on the date of
grant and is recorded as compensation expense ratably over the applicable service period, which is generally four years. Unvested restricted
stock units and vested and unvested stock options are forfeited in the event of termination of employment.
Income
Taxes
Income
taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences
attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective
tax bases, as well as for operating losses and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted
tax rates expected to be applied to taxable income in the years in which those temporary differences are expected to be recovered or
settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes
the enactment date. Deferred tax assets and liabilities are recorded net as long-term on the consolidated balance sheets.
Foreign Currency
Assets and liabilities of non-U.S. operations where
the functional currency is other than the U.S. dollar are translated from the functional currency into U.S. dollars at year end exchange
rates, and revenues and expenses are translated at average rates prevailing during the year. Resulting translation adjustments are accumulated
as part of accumulated other comprehensive income. Transaction gains or losses are recognized in income or loss in the period in which
they occur. The cumulative translation adjustment for the year ended December 31, 2023 was less than $1,000 and therefore not separately
reported on the consolidated financial statements.
A
valuation allowance is recorded when it is more likely than not that some or all of the net deferred tax assets will not be realized.
Accordingly, the Company provides a valuation allowance, if necessary, to reduce net deferred tax assets to the amount that is expected
to be realized.
Tax
positions taken or expected to be taken in the course of preparing the Company’s tax returns are required to be evaluated to determine
whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authority. Tax positions not
deemed to meet a “more-likely-than-not” threshold would be recorded as a tax expense in the current year.
When
necessary, the Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
Net
Loss per Share
Basic
net loss per share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of shares outstanding
during the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by adjusting the weighted-average
number of shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock
method. For purposes of the diluted net loss per share calculation, warrants to purchase common stock and stock options are considered
to be common stock equivalents, but have been excluded from the calculation of diluted net loss per share, as their effect would be anti-dilutive
for all periods presented. Therefore, basic and diluted net loss per share applicable to common stockholders were the same for all periods
presented.
Warrant
Liability
The
Company classifies warrants to purchase shares of its common stock as a liability on its consolidated balance sheets when the warrant
is a free-standing financial instrument that may require the Company to transfer cash consideration upon exercise and that cash transfer
event would be out of the Company’s control. Such a “liability warrant” is initially recorded at fair value on date
of grant using the Black-Scholes model and net of issuance costs, and it is subsequently re-measured to fair value at each subsequent
balance sheet date. Changes in the fair value of the warrant are recognized as a component of other income (expense), net in the consolidated
statements of operations. The Company will continue to adjust the liability for changes in fair value until the earlier of the exercise
or expiration of the warrant.
For
warrants that do not meet the criteria of a liability warrant and are classified on the Company’s consolidated balance sheets as
equity instruments, the Company uses the Black-Scholes model to measure the value of the warrants at issuance and then applies the relative
fair-value of the equity transaction between common stock, preferred stock and warrants. Common stock and equity-classified warrants
each are considered permanent equity.
Concentration
of Credit Risk
Financial
investments that potentially subject the Company to credit risk consist of cash. The Company has all cash at accredited financial institutions.
Bank accounts in the United States are insured by the Federal Deposit Insurance Corporation (FDIC) up to $250,000. The Company does not
believe that it is subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
Recent
Accounting Pronouncements
From
time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies we adopt as of the specified effective
date. Unless otherwise discussed below, we do not believe that the adoption of recently issued standards have or may have a material
impact on our consolidated financial statements.
In
June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial
Instruments (ASU 2016-12). The new standard requires that expected credit losses relating to financial assets measured on an amortized
cost basis and available-for-sale debt securities be recorded through an allowance for credit losses. It also limits the amount of credit
losses to be recognized for available-for-sale debt securities to the amount by which carrying value exceeds fair value and also requires
the reversal of previously recognized credit losses if fair value increases. The Company adopted this standard on January 1, 2023, and
the adoption of ASU 2016-13 did not have a material impact on its consolidated financial statements.
3.
Fair Value Measurements
Fair
value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal
or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date.
The
Company utilizes a valuation hierarchy for disclosure of the inputs to the valuations used to measure fair value. This hierarchy prioritizes
the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or
liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for
the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial
instrument. Level 3 inputs are unobservable inputs based on the Company’s own assumptions used to measure assets and liabilities
at fair value. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input
that is significant to the fair value measurement.
The Company had no assets or liabilities classified as Level 2 or Level
3 as of December 31, 2023 and 2022. In 2023, the Company had a certificate of deposit which matured in October 2023 with the remaining
$1.2 million released from short-term investments into cash and cash equivalents. The carrying value of financial instruments (consisting
of cash, accounts payable, accrued compensation and accrued expenses) is considered to be representative of their respective fair values
due to the short-term nature of those instruments.
Investment
income is included as interest income in the accompanying consolidated statement of operations for the year ended December 31, 2023.
There
were no transfers between Level 1, Level 2 and Level 3 in either of the years ended December 31, 2023 and December 31, 2022.
4.
Prepaid Expenses and Other Current Assets
Prepaid
expenses and other current assets consist of the following:
Schedule
of Prepaid expenses and Other Current Assets
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Deposits | |
$ | — | | |
$ | 20 | |
| Insurance | |
| 8 | | |
| 8 | |
| Prepaid contracts | |
| 79 | | |
| 51 | |
| Total prepaid expenses and other current assets | |
$ | 87 | | |
$ | 79 | |
5.
Property, Plant and Equipment, Net
Property,
plant and equipment, net consist of the following:
Schedule
of Property Plant and Equipment Net
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Leasehold improvements | |
$ | 35 | | |
$ | 35 | |
| Furniture, machinery and equipment | |
| 1,414 | | |
| 1,405 | |
| Computer equipment and software | |
| 38 | | |
| 36 | |
| Total property, plant and equipment | |
| 1,487 | | |
| 1,476 | |
| Less: accumulated depreciation | |
| (1,462 | ) | |
| (1,427 | ) |
| Property, plant and equipment, net | |
$ | 25 | | |
$ | 49 | |
Depreciation
expense amounted to approximately $35,000 and $52,000 for the years ended December 31, 2023 and 2022, respectively.
6. Long-term prepaid contracts
We have contracted with partners
relating to our clinical trial activities. Upon execution of the contracts, we made initial payments of $1.2
million as deposits recorded as long-term assets and will be applied against final invoices which are more than a year away. The deposits
will be recorded as expense when the clinical trial is substantially completed. Costs for the clinical trial activities throughout
our clinical trial under these contracts are recognized as expense and payable based on costs incurred.
7.
Accrued and Other Current Liabilities
Accrued
and other current liabilities consist of the following:
Schedule
of Accrued and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Advisory costs | |
$ | 325 | | |
$ | 300 | |
| Legal costs | |
| — | | |
| 135 | |
| Audit services | |
| 70 | | |
| 80 | |
| Payroll | |
| 79 | | |
| 55 | |
| Other liabilities | |
| 1 | | |
| 12 | |
| Total expenses | |
$ | 475 | | |
$ | 582 | |
8.
Warrant Liability
During
2016 and 2017, the Company closed a sale of shares of the Company’s common stock, the issuance of warrants to purchase shares of
common stock, and the issuance of warrants to the placement agent for each transaction. Due to a cash put provision within the warrant
agreement, which could be enacted in certain change in control events, a liability associated with those 1,044,396 warrants were initially
recorded at fair value and subsequently re-measured each reporting period. The changes in the fair value between issuance and the end
of each reporting period is recorded as a component of other income (expense), net in the consolidated statements of operations.
During
2017, the holders of 952,184 warrants agreed to a modification of the term which removed the cash put provision. The remaining 92,212
warrants continued to be re-measured at each reporting period as long as they were outstanding and un-modified. In February 2022, the
remaining 92,212 warrants expired unexercised.
The
following table presents a reconciliation of the Company’s warrant liabilities for the year ended December 31, 2022:
Schedule
of Warrant Liability
| |
|
Warrant
Liability |
|
| |
|
(in
thousands) |
|
| Balance
as of January 1, 2022 |
|
$ |
2 |
|
| Change
in fair value upon re-measurement |
|
|
(2 |
) |
| Balance
as of December 31, 2022 |
|
$ |
— |
|
9.
Commitments and Contingencies
On
April 14, 2017, representatives for the estate of an individual plaintiff filed a wrongful death complaint with the Suffolk Superior
Court, in the County of Suffolk, Massachusetts, against the Company and other defendants, including Harvard Bioscience, Inc., or HBIO,
the former parent of the Company that spun off the Company in 2013, as well as another third party. The complaint sought payment for
an unspecified amount of damages and alleged that the plaintiff sustained terminal injuries allegedly caused by products provided by
certain of the named defendants and utilized in connection with surgeries performed by third parties in Europe in 2012 and 2013. This
lawsuit related to the Company’s first-generation trachea scaffold technology for which the Company discontinued development in
2014, and not to the Company’s current HRGN Esophageal Implant.
On
April 27, 2022, the Company and HBIO executed a settlement with the plaintiffs (the “Settlement”), which resolves all claims
relating to the litigation. The Settlement resulted in the dismissal with prejudice of the wrongful death claim, and neither the Company
nor HBIO admit any fault or liability in connection with the claim. The Settlement also resolved any and all claims by and between the
parties and the Company’s product liability insurance carriers, which resulted in the dismissal with prejudice of all claims asserted
by or against those carriers, the Company and HBIO.
In
relation to the litigation, the Company paid approximately $5.9 million of aggregate costs related to the lawsuit. As of December 31,
2022, all such lawsuit related costs had been paid or otherwise satisfied as provided below. This aggregate amount included the cost
of legal and related costs incurred by the Company, which consisted of attorneys’ fees and advisor and specialist costs as part
of its defense in this matter. On March 3, 2022, the Company received a cash payment of approximately $0.1 million from Medmarc, the
Company’s insurance carrier. This amount represented a reimbursement of previously incurred legal costs and was recorded as a reduction
to general and administrative expenses during the year ended December 31, 2022.
With
respect to such $5.9 million of costs described above, the Company was required to either pay such costs directly or indemnify HBIO as
to such amounts it incurs. Of such amounts, the Company anticipated that HBIO would pay an aggregate amount of $4.0 million by the end
of the second quarter of 2022. With respect to the indemnification obligation of the Company to HBIO pertaining to such costs, the Company
and HBIO entered into a Preferred Issuance Agreement dated as of April 27, 2022 (the PIA). In connection with the PIA, the Company and
HBIO agreed that once HBIO had paid at least $4.0 million in such costs, to satisfy the Company’s indemnification obligations with
respect thereto, in lieu of paying cash, the Company would issue senior 8% convertible preferred stock to HBIO that will contain terms
as described in the PIA, including the term sheet attached thereto. On June 10, 2022, following the execution of a subscription agreement
and HBIO providing evidence of payment of the requisite $4.0 million amount, the Company issued HBIO 4,000 shares of Series E 8% Convertible
Preferred Stock at a price of $1,000 per share to satisfy the Company’s related indemnification obligations aggregating $4.0 million,
which included the accrual for contingency of $3.3 million and approximately $0.8 million of legal and related costs paid on behalf of
the Company by HBIO previously included in accrued expenses.
From
time to time, the Company may be involved in various claims and legal proceedings arising in the ordinary course of business. Other than
the above matter, there are no such matters pending that the Company expects to be material in relation to its business, financial condition,
results of operations, or cash flows.
We currently have a co-development initiative
with Yale University and the McGowan Institute for Regenerative Medicine at the University of Pittsburgh. We are required to make advance
payments of approximately $130,000 and $61,000, respectively at inception of the contracts. We plan to make these advance payments in
the second quarter of 2024. The universities started preparatory work in 2023 with substantial work to be done in 2024. Either party
can terminate the contract with reasonable notice and any incurred costs will be reimbursed by us to the universities.
10.
Leases
The
Company leases laboratory and office space and certain equipment with remaining terms ranging from 1 year to 3 years.
The
laboratory and office space arrangement is under a sublease that was renewed in December of 2022 and currently extends through May 31,
2024. This lease automatically renews annually for one-year periods unless the Company or the counterparty provides a notice of termination
within one hundred and eighty days prior to May 31st of each year.
All
of the Company’s leases qualify as operating leases. The following table summarizes the presentation of the Company’s operating
leases in its consolidated balance sheets:
Schedule of Operating Leases in Consolidated Balance Sheets
| | |
| |
December 31, | |
| | |
Balance Sheet Classification | |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Assets: | |
| |
| | |
| |
| Operating lease assets | |
Right-of-use asset, net | |
$ | 48 | | |
$ | 147 | |
| Liabilities: | |
| |
| | | |
| | |
| Current portion of operating lease liabilities | |
Current portion of operating lease liabilities | |
| 48 | | |
| 99 | |
| Operating lease liabilities, net of current portion | |
Operating lease liabilities, net of current portion | |
| — | | |
| 48 | |
| Total operating lease liabilities | |
| |
$ | 48 | | |
$ | 147 | |
Cash
paid for leases during each
of the years ended December 31, 2023 and 2022 amounted to approximately $127,000 and $121,000, respectively.
The
weighted average remaining lease terms and weighted average discount rates as of December 31, 2023 and 2022 were as follows:
Schedule
of Weighted Average Lease Term and Discount Rates
| | |
Year ended December 31, | |
| | |
2023 | | |
2022 | |
| Remaining lease term (in years) | |
| 0.48 | | |
| 1.43 | |
| Discount rate | |
| 14.66 | % | |
| 14.74 | % |
The
following table summarizes the effect of lease costs in the Company’s consolidated statements of operations:
Schedule
of Operating Lease Expense Categories in Consolidated Statements of Operations
| | |
| |
For the Year Ended December 31, | |
| | |
| |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Operating lease expense | |
Research and development | |
$ | 68 | | |
$ | 77 | |
| | |
Sales and marketing | |
| 15 | | |
| — | |
| | |
General and administrative | |
| 44 | | |
| 44 | |
| | |
Total | |
$ | 127 | | |
$ | 121 | |
The
minimum lease payments for the next year is as follows:
Schedule
of Minimum Lease Payments
| | |
As of | |
| | |
December 31, 2023 | |
| | |
(in thousands) | |
| 2024 | |
$ | 50 | |
| Total lease payments | |
| 50 | |
| Less: imputed interest | |
| (2 | ) |
| Present value of operating lease liabilities | |
$ | 48 | |
11.
Income Taxes
A
reconciliation of taxes utilizing the expected federal tax rate of 21% and the effective tax rate is as follows:
Schedule of Effective Income Tax
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| Computed “expected” income tax benefit | |
| 21.0 | % | |
| 21.0 | % |
| State income tax benefit, net of federal income tax benefit | |
| 6.3 | % | |
| 6.3 | % |
| Tax credits | |
| 0.6 | % | |
| 0.8 | % |
| Change in valuation allowance | |
| (27.9 | )% | |
| (28.1 | )% |
| Total income taxes | |
| — | % | |
| — | % |
The
components of the Company’s deferred tax assets and liabilities are as follows:
Schedule
of Deferred tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Deferred tax assets: | |
| | | |
| | |
| Operating loss and credit carryforwards | |
$ | 19,167 | | |
$ | 20,487 | |
| Capitalized research and development | |
| 1,288 | | |
| 1,083 | |
| Stock-based compensation | |
| 2,504 | | |
| 1,566 | |
| Lease liabilities | |
| 13 | | |
| 40 | |
| Total deferred tax assets | |
| 22,972 | | |
| 23,176 | |
| Less: valuation allowance | |
| (22,959 | ) | |
| (23,136 | ) |
| Deferred tax assets | |
| 13 | | |
| 40 | |
| | |
| | | |
| | |
| Deferred tax liability: | |
| | | |
| | |
| Operating lease assets | |
| (13 | ) | |
| (40 | ) |
| Total deferred tax liability | |
| (13 | ) | |
| (40 | ) |
| Deferred
Tax Net | |
$ | — | | |
$ | — | |
The
Company has recorded a valuation allowance against its deferred tax assets for the years ended December 31, 2023 and 2022, because the
Company’s management believes that it is more likely than not that these assets will not be realized. The valuation allowance decreased
by approximately $0.2
million for the year ended December 31, 2023 and increased by approximately $2.9 million
for the year ended December 31, 2022, respectively, primarily as a result of operating losses generated with no corresponding financial
statement benefit.
As
of December 31, 2023, the Company had federal net operating loss carryforwards, or NOLs, of approximately $68.7 million to offset future
federal taxable income and state NOLs of approximately $68.1 million to offset future state taxable income. The federal and state NOLs
generated for annual periods prior to January 1, 2018 begin to expire in 2033. The Company’s federal NOL generated for the years
ended December 31, 2018 through December 31, 2023, which amount to $42.3 million, can be carried forward indefinitely, however, are limited
to be utilized to offset 80% of taxable income in each successive year. As of December 31, 2023, the Company also has federal and state
tax research and development credit carryforwards of approximately $1.5 million and $1.0 million, respectively, to offset future income
taxes. The federal and state research and development tax credit carryforwards begin to expire in 2033 and 2029, respectively.
Under
the provisions of the Internal Revenue Code, the net operating loss and tax credit carryforwards are subject to review and possible adjustment
by the Internal Revenue Service and state tax authorities. Net operating loss and tax credit carryforwards may become subject to an annual
limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period
in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code, respectively, as well as similar state provisions.
This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount
of the annual limitation is determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership
changes may further affect the limitation in future years. The Company has recently completed several equity financings transactions
which have either individually or cumulatively resulted in a change in control as defined by Sections 382 and 383 of the Internal Revenue
Code or could result in a change in control in the future. The Company does not believe the impact of any limitation on the use of its
net operating loss or credit carryforwards will have a material impact on the Company’s consolidated financial statements since
the Company has a full valuation allowance against its net deferred tax assets due to the uncertainty regarding future taxable income
for the foreseeable future.
For
all years through December 31, 2023, the Company generated research credits but has not conducted a study to document the qualified activities.
This study may result in an adjustment to the Company’s research and development credit carryforwards; however, until a study is
completed, and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been
provided against the Company’s research and development credits and, if an adjustment is required, this adjustment would be offset
by an adjustment to the deferred tax asset established for the research and development credit carryforwards and the valuation allowance.
Harvard
Bioscience received a Supplemental Ruling to the Private Letter Ruling dated March 22, 2013 from the IRS to the effect that, among other
things, the Separation and Distribution by Harvard Bioscience will qualify as a transaction that is tax-free for U.S. federal income
tax purposes under Section 355 and 368(a)(1)(D) of the Internal Revenue Code continuing in effect. The private letter and supplemental
rulings and the tax opinion that Harvard Bioscience received from legal counsel to Harvard Bioscience rely on certain representations,
assumptions and undertakings, including those relating to the past and future conduct of the Harvard Apparatus Regenerative Technology
business, and neither the private letter and supplemental rulings nor the opinion would be valid if such representations, assumptions
and undertakings were incorrect. Moreover, the private letter and supplemental rulings do not address all the issues that are relevant
to determining whether the Distribution will qualify for tax-free treatment. Notwithstanding the private letter and supplemental rulings
and opinion, the IRS could determine the Distribution should be treated as a taxable transaction for U.S. federal income tax purposes
if, among other reasons, it determines any of the representations, assumptions or undertakings that were included in the request for
the private letter and supplemental rulings are false or have been violated or if it disagrees with the conclusions in the opinion that
are not covered by the IRS ruling.
To
preserve the tax-free treatment to Harvard Bioscience of the Separation and Distribution, for the two-year period following the Distribution,
which such period ended November 1, 2015, the Company was limited, except in specified circumstances, from entering into certain transactions
pursuant to which all or a portion of the Company’s stock would be acquired, whether by merger or otherwise; issuing equity securities
beyond certain thresholds; repurchasing the Company’s common stock; and ceasing to actively conduct the Company’s regenerative
medicine business. In addition, at all times, including during and following such two-year period, the Company may not take or fail to
take any other action that prevents the Separation and Distribution and related transactions from being tax-free.
If
the Distribution fails to qualify for tax-free treatment, in general, Harvard Bioscience would be subject to tax as if it had sold the
Company’s common stock in a taxable sale for its fair market value, and Harvard Bioscience stockholders who received shares of
Harvard Apparatus Regenerative Technology common stock in the Distribution would be subject to tax as if they had received a taxable
Distribution equal to the fair market value of such shares.
Under
the tax sharing agreement between Harvard Bioscience and the Company, the Company would generally be required to indemnify Harvard Bioscience
against any tax resulting from the Distribution to the extent that such tax resulted from (i) an acquisition of all or a portion of the
Company’s stock or assets, whether by merger or otherwise, (ii) other actions or failures to act by the Company, or (iii) any of
the Company’s representations or undertakings being incorrect or violated. The Company’s indemnification obligations to Harvard
Bioscience and its subsidiaries, officers and directors are not limited by any maximum amount. If the Company is required to indemnify
Harvard Bioscience or such other persons under the circumstances set forth in the tax sharing agreement, the Company may be subject to
substantial liabilities.
All
deferred tax assets prior to the Separation remained with Harvard Bioscience.
The
Company has determined that any uncertain tax positions would have no material impact on the consolidated financial statements of the
Company and there are no unrecognized tax benefits or related interest and penalties accrued for the period for the years ended December
31, 2023 and 2022.
The
Company is subject to U.S. federal income tax and Massachusetts state income tax. The statute of limitations for assessment by the IRS
and state tax authorities is open for all periods from inception through December 31, 2022; currently, no federal or state income tax
returns are under examination by the respective taxing authorities.
On
March 27, 2020, the Coronavirus Aid, Relief, and Economic Security, or CARES, Act was signed into law making several changes to the Internal
Revenue Code. The changes include but are not limited to increasing the limitation on the amount of deductible interest expense, allowing
companies to carryback certain net operating losses, and increasing the amount of net operating loss carryforwards that corporations
can use to offset taxable income. The tax law changes in the CARES Act did not have a material impact on the Company’s income tax
provision.
12.
Employee Benefit Plan
The
Company sponsors a retirement plan for its U.S. employees, which includes an employee savings plan established under Section 401(k) of
the U.S. Internal Revenue Code, or the 401(k) Plan. The 401(k) Plan covers substantially all full-time employees who meet certain eligibility
requirements. Contributions to the retirement plan are at the discretion of management. The Company’s matching contributions to
the plan were approximately $66,000 and $35,000 for the years ended December 31, 2023 and 2022, respectively.
13.
Series E Convertible Preferred Stock
On
April 28, 2022, the Company entered into a Preferred Issuance Agreement, or PIA, with Harvard Bioscience, Inc., or HBIO, dated as of
April 27, 2022. Pursuant to the PIA, the Company and HBIO agreed that once HBIO has paid at least $4.0 million in certain settlement
and related legal expenses, to satisfy the Company’s indemnification obligations with respect thereto, in lieu of paying cash,
the Company would issue senior convertible preferred stock to HBIO that will contain terms as described in the PIA.
On
June 10, 2022, following the execution of a subscription agreement and HBIO providing evidence of payment of the requisite $4.0 million
amount, the Company issued HBIO 4,000 shares of Series E Convertible Preferred Stock, or Series E Preferred, at a price of $1,000 per
share to satisfy the Company’s related indemnification obligations pertaining to the $4.0 million, in lieu of paying cash.
On
January 18, 2023, HBIO converted 200 Series E Preferred Shares with accrued dividends of $9,545 into 31,933 shares of common stock.
In
connection with the private placement, as of April 12, 2023, the Company had received $6.0 million in aggregate proceeds in such private
placement. The private placement resulted in gross proceeds of at least $4.0 million which triggered the mandatory conversion of all
the Company’s outstanding Series E Preferred Stock and related accrued dividends into shares of common stock at a conversion price
of $6.00 per share. The conversion resulted in 674,693 shares of common stock being issued to the holder of the Series E Preferred Stock.
Following such conversion, there are no shares of Series E Preferred Stock outstanding.
There
were no
shares of any of the classes of preferred stock outstanding as of December 31, 2023. There were no changes to authorized shares for
the years ending December 31, 2022 and 2023. Authorized shares for each preferred stock class are as follows:
Schedule
of Categories of Preferred Stock
| | |
Authorized | |
| Undesignated Preferred Stock | |
| 979,000 | |
| Series B Convertible Preferred Stock | |
| 1,000,000 | |
| Series C Convertible Preferred Stock | |
| 4,000 | |
| Series D Convertible Preferred Stock | |
| 12,000 | |
| Series E Convertible Preferred Stock | |
| 5,000 | |
14.
Common Stock
The
Company has 60,000,000 shares authorized as of December 31, 2023 and 40,961,765 shares of common stock available for issuance.
The
following represent the Company’s common stock transactions during December 31, 2023 and 2022:
2023
Capital Transactions
On
April 12, 2023 and on March 31, 2023, the Company entered into Securities Purchase Agreements, each a Purchase Agreement, with new and
existing investors, the Investors, pursuant to which the Investors agreed to purchase in a private placement an aggregate of 1,000,967
shares of common stock for the aggregate purchase price of approximately $6 million with a purchase price per unit of $6.00.
2022
Capital Transactions
On
May 12, 2022, the Company entered into Securities Purchase Agreements, each a Purchase Agreement, with new and existing investors, the
Investors, pursuant to which the Investors agreed to purchase in a private placement an aggregate of 854,771 shares of common stock and
warrants to purchase 427,390 shares of common stock, subject to adjustment as provided in the warrant agreement, the Warrants, for the
aggregate purchase price of approximately $5.1 million with a purchase price per unit of $5.92, the Private Placement. Each unit consisted
of one share of common stock and a warrant to purchase one half of one share of common stock, subject to adjustment, as provided in the
Warrants. The Company received an aggregate of $5.1 million gross and net proceeds from the Private Placement by May 16, 2022.
The
$5.1 million of gross and net proceeds were allocated $3.6 million and $1.5 million to the common stock and warrants, respectively.
The Company classified these warrants on its consolidated balance sheets as equity as the warrants do not have any redemption features
nor a right to put for cash that is outside the control of the Company, and valued using the Black-Scholes model based on the following
weighted average assumptions:
Schedule
of Black-Scholes Model Based on Weighted Average Assumptions
| Risk-free interest rate | |
| 2.81 | % |
| Expected volatility | |
| 127.36 | % |
| Expected term | |
| 5 years | |
| Expected dividend yield | |
| — | |
| Exercise price | |
$ | 8.88 | |
| Market value of common stock | |
$ | 5.50 | |
In
June 2022, the Company issued 4,000 shares of Series E Convertible Preferred Stock at a price of $1,000 per share to satisfy certain
indemnification obligations in the amount of $4.0 million, in lieu of paying cash. The Company issued an aggregate of 180 shares of Series
E Convertible Preferred Stock relating to accrued dividends during the year ended December 31, 2022.
Warrant
to purchase common stock activity for the year ended December 31, 2022 was as follows:
Schedule of Warrant to Purchase Common Stock
| | |
| | |
Weighted-average | |
| | |
Amount | | |
exercise price | |
| Outstanding at January 1, 2022 | |
| 2,501,419 | | |
$ | 4.35 | |
| Issued | |
| 427,390 | | |
| 8.88 | |
| Exercised | |
| (775,000 | ) | |
| 7.17 | |
| Expired | |
| (1,040,187 | ) | |
| 7.59 | |
| Outstanding at December 31, 2022 | |
| 1,113,622 | | |
| 4.69 | |
| Outstanding at December 31, 2023 | |
| 1,113,622 | | |
| 4.69 | |
There was no warrant activity during the year ended December 31, 2023.
Employee
Stock Purchase Plan
The
Company maintains the 2013 Employee Stock Purchase Plan, or the ESPP Plan, whereas participating employees can authorize the Company
to withhold a portion of their base pay during consecutive six-month payment periods for the purchase of shares of the Company’s
common stock. At the conclusion of the period, participating employees can purchase shares of the Company’s common stock at 85%
of the lower of the fair market value of the Company’s common stock at the beginning or end of the period. Shares are issued under
the plan for the six-month periods ending June 30 and December 31. Under this plan, 7,500 shares of common stock are authorized for issuance
of which 4,534 shares have been issued as of December 31, 2023. There are 2,966 shares available for issuance as of December 31, 2023
and December 31, 2022. There was no ESPP Plan activity in 2023 or 2022.
15.
Share-based Compensation
Harvard
Apparatus Regenerative Technology Amended and Restated Equity Incentive Plan
The
Company maintains the Amended and Restated Equity Incentive Plan, or the Plan, for the benefit of certain officers, employees, non-employee
directors, and other key persons (including consultants and advisory board members). All options and awards granted under the Plan consist
of the Company’s shares of common stock. The Company’s policy is to issue stock available from its registered but unissued
stock pool through its transfer agent to satisfy stock option exercises and the vesting of restricted stock units. The vesting period
for awards is generally four years and the contractual life is ten years. Canceled and forfeited options and awards are available to
be reissued under the Plan.
As
of December 31, 2023, the Company’s Plan has 9,098,000
authorized shares to be issued under the Plan. There are 5,034,760
shares available for issuance under the Plan as of December 31, 2023.
Stock
option activity under the Plan for the years ended December 31, 2022 and 2023 was as follows:
Schedule of Stock Option Activity
| | |
Amount | | |
Weighted-average exercise price | | |
Weighted-average contractual life (years) | | |
Aggregate intrinsic value (in thousands) | |
| Outstanding at January 1, 2022 | |
| 2,332,603 | | |
$ | 3.93 | | |
| 8.30 | | |
$ | 294 | |
| Granted | |
| 334,418 | | |
| 4.84 | | |
| | | |
| | |
| Canceled / forfeited | |
| (150,097 | ) | |
| 3.39 | | |
| | | |
| | |
| Outstanding at December 31, 2022 | |
| 2,516,924 | | |
| 3.95 | | |
| 7.68 | | |
| 6,917 | |
| Granted | |
| 2,130,007 | | |
| 5.81 | | |
| | | |
| | |
| Exercised | |
| (65,264 | ) | |
| 2.29 | | |
| | | |
| | |
| Canceled / forfeited | |
| (604,378 | ) | |
| 6.13 | | |
| | | |
| | |
| Outstanding at December 31, 2023 | |
| 3,977,289 | | |
$ | 4.64 | | |
| 7.7 | | |
$ | 5,728 | |
| Options exercisable at December 31, 2023 | |
| 2,281,655 | | |
$ | 4.64 | | |
| 7.24 | | |
$ | 4,209 | |
| Options vested or expected to vest at December 31, 2023 | |
| 3,877,871 | | |
$ | 4.69 | | |
| 7.7 | | |
$ | 5,522 | |
The
Company’s outstanding stock options include 773,195 performance-based awards that have vesting provisions subject to the achievement
of certain business milestones. Total unrecognized compensation expense for the remaining performance-based awards is approximately $2.8
million. No expense has been recognized for these awards as of December 31, 2023 given that the milestone achievements for these awards
have not yet been deemed probable for accounting purposes.
Aggregate
intrinsic value for outstanding options for the year ended December 31, 2023 was approximately $5.7
million and calculated as the difference between the Company’s closing stock price of $4.99
per share as of December 29, 2023 and the weighted average exercise price of $4.64.
As of December 31, 2023, unrecognized compensation cost related to unvested non-performance-based awards amounted to $3.9
million, which will be recognized over a weighted-average period of 2.2
years.
The
weighted average assumptions for valuing the Company’s stock options granted were as follows:
Schedule of Weighted Average Assumptions
| | |
Year Ended December 31, | |
| | |
2023 | | |
2022 | |
| Risk-free interest rate | |
| 3.82 | % | |
| 2.74 | % |
| Expected volatility | |
| 125.35 | % | |
| 123.61 | % |
| Expected term (in years) | |
| 5.8 years | | |
| 5.8 years | |
| Expected dividend yield | |
| — | % | |
| — | % |
The
grant date fair value of stock options is estimated using the Black-Scholes option pricing model that takes into account the fair value
of its common stock, the exercise price, the expected life of the option, the expected volatility of its common stock, expected dividends
on its common stock, and the risk-free interest rate over the expected life of the option. The risk-free interest rate assumption is
based upon observed treasury bill interest rates (risk-free) appropriate for the expected term of the Company’s employee stock
options. The computation of expected volatility is based on the historical volatility of the Company’s common stock. The simplified
method of estimating expected term was used. The Company has not paid and do not anticipate paying cash dividends on the Company’s
shares of common stock; therefore, the expected dividend yield is assumed to be zero.
The
weighted average estimated fair value of stock options granted using the Black-Scholes model was $5.12 and $4.23 per share for the years
ended December 31, 2023 and 2022, respectively.
The
Company also estimated the fair value of non-employee share options using the Black-Scholes option pricing model reflecting the same
assumptions as applied to employee and director options in each of the reporting periods, other than the expected life, which is assumed
to be the remaining contractual life of the options.
Share-based
compensation expense related to the Plan for the years ended December 31, 2023 and 2022 was allocated as follows:
Schedule
of Share-based Compensation Expense
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Research and development | |
$ | 327 | | |
$ | 284 | |
| Selling, general and administrative | |
| 3,134 | | |
| 747 | |
| Total stock-based compensation | |
$ | 3,461 | | |
$ | 1,031 | |
16.
Net Loss per Share
Basic
and diluted net loss per share was calculated as follows:
Schedule of Basic and Diluted Net Loss Per Share
| | |
2023 | | |
2022 | |
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands, except shares and per share data) | |
| Net loss | |
$ | (8,945 | ) | |
$ | (6,073 | ) |
| Preferred stock dividends | |
| (77 | ) | |
| (180 | ) |
| Net loss attributable to common stockholders | |
| (9,022 | ) | |
| (6,253 | ) |
| Basic and diluted weighted average common shares outstanding | |
| 13,455,666 | | |
| 11,349,610 | |
| Basic and diluted net loss per share attributable to common stockholders | |
$ | (0.67 | ) | |
$ | (0.55 | ) |
The
Company’s potentially dilutive securities, which include stock options, unvested restricted common stock units and warrants, have
been excluded from the computation of diluted net loss per share whenever the effect of including them would be to reduce the net loss
per share. In periods where there is a net loss, the weighted average number of common shares outstanding used to calculate both basic
and diluted net loss per share attributable to common stockholders is the same.
The
following potential common shares were excluded from the calculation of diluted net loss per share attributable to common stockholders
for the years ended December 31, 2023 and 2022 because including them would have had an anti-dilutive effect:
Schedule
of Antidilutive Securities Excluded from Computation of Earnings per Share
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| Warrants to purchase common stock | |
| 1,113,622 | | |
| 1,113,622 | |
| Options to purchase common stock | |
| 3,977,289 | | |
| 2,516,924 | |
| Series E convertible preferred stock | |
| — | | |
| 619,259 | |
| Total | |
| 5,090,911 | | |
| 4,249,805 | |
17.
Segments and Geographical Information
The
Company’s chief operating decision maker is its Chief Executive Officer. The Company’s chief operating decision maker evaluates
the operating results of the Company’s reportable segments based on revenues and net income (loss).
The Company has two operating and reportable segments: i) Regenerative Biotech
focused on the development of regenerative medicine treatments with operations currently in the United States and ii) Longevity Products
relating to longevity products with operations currently in Asia. The following table presents the Company’s reportable
segment results for the year ended 2023:
Schedule of Reportable Segments
| | |
| | | |
| | |
|
| | |
| | |
Regenerative Biotech | | |
Longevity Products | |
|
Total | |
| 2023 | |
| | | |
| | |
|
| | |
| Revenues | |
$ | — | | |
$ | 103 | |
|
$ | 103 | |
| Net loss | |
| (8,677 | ) | |
| (268 | ) |
|
| (8,945 | ) |
| Total assets | |
| 2,426 | | |
| 188 | |
|
| 2,614 | |
18.
Subsequent Events
In March 2024, the Company received cash deposits
in escrow of approximately $0.3 million from a group of prospective investors pertaining to a potential private placement transaction.
These funds remain the respective investor’s property and are being held by the Company in its bank account with Bank of America
until the execution of a common stock purchase agreement.
On
February 1, 2024, the Company entered into a loan arrangement with Junli He, the Chairman and Chief Executive Officer of the Company
(the “Lender”), pursuant to which the Lender has agreed to loan the Company an aggregate amount of $500,000 as evidenced
by a Bridge Note executed by the Company in favor of, and accepted by, the Lender (the “Bridge Note”).
The
Bridge Note accrues interest at an annual fixed rate of 8%, and the principal amount thereof will be due and payable in full, together
with all accrued and unpaid interest thereon, on the earlier to occur of a) the closing date (or later date of capital being provided
pertaining to such continued offering that the following threshold is tripped) of the Company’s next capital raise that includes
gross proceeds of at least $5,000,000 or b) February 1, 2025. The Bridge Note provides for optional conversion at the discretion of the
Lender, contains covenants, and provides for certain events of default including if the Company fails to pay when due any amount owed
thereunder, fails to comply with any agreement, covenant, condition, provision or term contained therein and other customary events of
default.
Item
16. Form 10-K Summary.
None.
EXHIBIT
INDEX
The
following exhibits are filed as part of this Annual Report on Form 10-K. Where such filing is made by incorporation by reference to a
previously filed document, such document is identified.
Exhibit
Number |
|
Description
of Exhibit |
| 2.1§ |
|
Separation and Distribution Agreement between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc. dated as of October 31, 2013 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on November 6, 2013, and incorporated by reference thereto). |
| 3.1 |
|
Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 3.2 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. dated March 30, 2016 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on March 31, 2016, and incorporated by reference thereto). |
| 3.3 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. dated May 26, 2016 (previously filed as an exhibit to the Company’s Annual Report on Form 10-K, filed on March 17, 2017, and incorporated by reference thereto). |
| 3.4 |
|
Certificate of Designations, Preferences and Rights of Series A Preferred Stock of Harvard Apparatus Regenerative Technology, Inc. classifying and designating the Series A Junior Participating Cumulative Preferred Stock (previously filed as an exhibit to the Company’s Registration Statement on Form 8-A, filed October 31, 2013, and incorporated by reference thereto). |
| 3.5 |
|
Certificate of Designation of Series B Convertible Preferred Stock of Harvard Apparatus Regenerative Technology, Inc. classifying and designating the Series B Convertible Preferred Stock (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on February 12, 2015, and incorporated by reference thereto). |
| 3.6 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. dated April 26, 2017 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on April 27, 2017, and incorporated by reference thereto). |
| 3.7 |
|
Certificate of Designations, Preferences, Rights and Limitations of Series C Convertible Preferred Stock of Harvard Apparatus Regenerative Technology, Inc. classifying and designating the Series C Convertible Preferred Stock (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on August 17, 2017, and incorporated by reference thereto). |
| 3.8 |
|
Certificate of Elimination of Series A Junior Participating Cumulative Preferred Stock (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on August 17, 2017, and incorporated by reference thereto). |
| 3.9 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. dated December 22, 2017 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on December 22, 2017, and incorporated by reference thereto). |
| 3.10 |
|
Certificate
of Designations, Preferences, Rights and Limitations of Series D Convertible Preferred Stock of Harvard Apparatus Regenerative Technology,
Inc. classifying and designating the Series D Convertible Preferred Stock (previously filed as an exhibit to the Company’s
Current Report on Form 8-K, filed on January 3, 2018, and incorporated by reference thereto). |
| 3.11 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation of Harvard Apparatus Regenerative Technology, Inc. dated May 24, 2019 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on May 28, 2019, and incorporated by reference thereto). |
| 3.12 |
|
Amended and Restated By-laws of the Harvard Apparatus Regenerative Technology, Inc. (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on March 31, 2016, and incorporated by reference thereto). |
| 3.13 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation (previously filed as an exhibit to the Current Report on Form 8-K, filed on July 20, 2023, and incorporated herein by reference). |
| 3.14 |
|
Third Amended and Restated Bylaws (previously filed as an exhibit to the Current Report on Form 8-K, filed on July 20, 2023, and incorporated herein by reference). |
| 4.1 |
|
Specimen Stock Certificate evidencing shares of common stock (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 4.2 |
|
Specimen Series B Convertible Preferred Stock Certificate (previously filed as an exhibit to the Company’s Annual Report on Form 10-K, filed on March 27, 2015, and incorporated by reference thereto). |
| 4.3 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on January 3, 2018, and incorporated by reference thereto). |
| 4.4 |
|
Form of Amendment to Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on December 18, 2019, and incorporated by reference thereto). |
| 4.5 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on January 2, 2020, and incorporated by reference thereto). |
| 4.6 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8 K, filed on June 22, 2021, and incorporated by reference thereto). |
| 4.7 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8 K, filed on September 8, 2021, and incorporated by reference thereto). |
| 4.8 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8 K, filed on November 30, 2021, and incorporated by reference thereto). |
| 4.9 |
|
Description of Securities (previously filed as an exhibit to the Company’s Annual Report on Form 10 K, filed on March 27, 2020, and incorporated by reference thereto). |
| 4.10 |
|
Form of Common Stock Purchase Warrant (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on May 13, 2022, and incorporated by reference thereto). |
| 10.1 |
|
Intellectual Property Matters Agreement between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc. dated as of October 31, 2013 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on November 6, 2013, and incorporated by reference thereto). |
| 10.2 |
|
Product Distribution Agreement between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc. dated as of October 31, 2013 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on November 6, 2013, and incorporated by reference thereto). |
| 10.3 |
|
Tax Sharing Agreement between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc. dated as of October 31, 2013 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on November 6, 2013, and incorporated by reference thereto). |
| 10.4 |
|
Sublease by and between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc. dated as of October 31, 2013 (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on November 6, 2013, and incorporated by reference thereto). |
| 10.5 |
|
Form of Indemnification Agreement for Officers and Directors (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.6#
|
|
Third Amended and Restated Equity Incentive Plan, as amended (previously filed as exhibit to the Company’s Quarterly Report on Form 10-Q, filed on November 13, 2023, and incorporated by reference thereto). |
| 10.7 |
|
Employee Stock Purchase Plan (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.8# |
|
Form of Incentive Stock Option Agreement (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.9# |
|
Form of Non-Qualified Stock Option Agreement for executive officers (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.10# |
|
Form of Non-Qualified Stock Option Agreement for directors (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.11# |
|
Form of Deferred Stock Award Agreement (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.12† |
|
Sublicense Agreement dated as of December 7, 2012 between Harvard Apparatus Regenerative Technology, Inc. and Harvard Bioscience, Inc., and related Trademark License Agreement, dated December 19, 2002, by and between Harvard Bioscience, Inc. and President and Fellows of Harvard College (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on March 7, 2023, and incorporated by reference thereto). |
| 10.13 |
|
Patent Rights Assignment dated December 21, 2012 between Harvard Apparatus Regenerative Technology, Inc. and Dr. Paolo Macchiarini (previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B, filed July 31, 2013, and incorporated by reference thereto). |
| 10.14# |
|
Offer Letter, dated June 4, 2018, between Harvard Apparatus Regenerative Technology, Inc. and William Fodor, PhD (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on July 10, 2018, and incorporated by reference thereto). |
| 10.15 |
|
Form of Securities Purchase Agreement (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on April 6, 2023 and incorporated herein by reference). |
| 10.16# |
|
Employment Agreement, dated August 8, 2022, between Harvard Apparatus Regenerative Technology, Inc. and Joseph L. Damasio, Jr. (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on August 9, 2022 and incorporated by reference thereto). |
| 10.17# |
|
Amended and Restated Employment Agreement, dated January 11, 2023, between Harvard Apparatus Regenerative Technology, Inc. and David Green (previously filed as an exhibit to the Company’s Current Report on Form 8-K, filed on January 12, 2023 and incorporated by reference thereto). |
| 10.18# |
|
Employment Agreement, effective as of March 1, 2023, by and between Harvard Apparatus Regenerative Technology, Inc. and Junli He (previously filed as an exhibit to the Current Report on Form 8-K, filed on March 14, 2023, and incorporated herein by reference). |
| 10.19# |
|
Amendment to Employment Agreement, dated as of July 10, 2023, by and between Harvard Apparatus Regenerative Technology, Inc. and Junli He (previously filed as an exhibit to the Current Report on Form 8-K, filed on July 10, 2023, and incorporated herein by reference). |
| 21.1* |
|
Subsidiaries of Harvard Apparatus Regenerative Technology, Inc. |
| 23.1* |
|
Consent of Marcum LLP. |
| 31.1* |
|
Certification of Chief Executive Officer of Harvard Apparatus Regenerative Technology, Inc., pursuant to Rules 13a-15(e) and 15d-15(e), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| 31.2* |
|
Certification of Chief Financial Officer of Harvard Apparatus Regenerative Technology, Inc., pursuant to Rules 13a-15(e) and 15d-15(e), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| 32.1** |
|
Certification of Chief Executive Officer of Harvard Apparatus Regenerative Technology, Inc., pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| 32.2** |
|
Certification of Chief Financial Officer of Harvard Apparatus Regenerative Technology, Inc., pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| 101.INS* |
|
Inline
XBRL Instance Document. |
| 101.SCH* |
|
Inline
XBRL Taxonomy Extension Schema Document. |
| 101.CAL* |
|
Inline
XBRL Taxonomy Extension Calculation Linkbase Document. |
| 101.DEF* |
|
Inline
XBRL Taxonomy Extension Definition Linkbase Document. |
| 101.LAB* |
|
Inline
XBRL Taxonomy Extension Label Linkbase Document. |
| 101.PRE* |
|
Inline
XBRL Taxonomy Extension Presentation Linkbase Document. |
| 104 |
|
Cover
Page Interactive Data File (formatted in iXBRL and contained in Exhibit 101) |
| * |
Filed
herewith. |
| |
|
| ** |
This
certification shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, or otherwise
subject to the liability of that section, nor shall it be deemed to be incorporated by reference into any filing under the Securities
Act of 1933 or the Securities Exchange Act of 1934. |
| |
|
| # |
Management
contract or compensatory plan or arrangement. |
| |
|
| § |
The
schedules and exhibits to the Separation and Distribution Agreement have been omitted. A copy of any omitted schedule or exhibit
will be furnished to the SEC supplementally upon request. The Company will furnish to stockholders a copy of any exhibit without
charge upon written request. |
| |
|
| † |
Certain
identified information has been excluded from the exhibit because it is both not material and is of the type that the registrant
treats as private or confidential. |
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed
on its behalf by the undersigned, thereunto duly authorized.
| |
Harvard
Apparatus Regenerative Technology, Inc. |
| Date:
March 28, 2024 |
|
|
| |
By:
|
/s/
Junli (Jerry) He |
| |
|
Junli
(Jerry) He |
| |
|
Chief
Executive Officer |
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the
registrant and in the capacities and on the dates indicated:
| Signature |
|
Title |
|
Date |
| |
|
|
|
|
| /s/
Junli (Jerry) He |
|
Chief
Executive Officer, Director, and Chairman |
|
|
| Junli
(Jerry) He |
|
(principal
executive officer) |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
Joseph Damasio Jr. |
|
Chief
Financial Officer |
|
|
| Joseph
Damasio Jr. |
|
(principal
financial officer and principal accounting officer) |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
Jason Jing Chen |
|
|
|
|
| Jason
Jing Chen |
|
Vice
Chairman |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
David Green |
|
|
|
|
| David
Green |
|
Director |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
Ting Li |
|
|
|
|
| Ting
Li |
|
Director |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
Ronald Packard |
|
|
|
|
| Ronald
Packard |
|
Director |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
Herman Sanchez |
|
|
|
|
| Herman
Sanchez |
|
Director |
|
March 28, 2024 |
| |
|
|
|
|
| /s/
James Shmerling |
|
|
|
|
| James
Shmerling |
|
Director |
|
March 28, 2024 |
Exhibit
21.1
Subsidiaries
of the Registrant
| Harvard
Apparatus Regenerative Technology Limited |
(China) |
| Harvard
Apparatus Regenerative Technology GmbH |
(Germany) |
| Harvard
Apparatus Regenerative Technology Limited |
(Hong
Kong) |
EXHIBIT
23.1
Consent
of Independent Registered Public Accounting Firm
We
consent to the incorporation by reference in the Registration Statement of Harvard Apparatus Regenerative Technology, Inc. (formerly
known as Biostage, Inc.) on Forms S-8 (No. 333-192026, 333-192027, 333-203105, 333-212993, 333-219988, 333-225336, 333-239346, and
333-275592) of our report dated March 28, 2024, which includes an explanatory paragraph as to the Company’s ability to
continue as a going concern, with respect to our audits of the consolidated financial statements of Harvard Apparatus Regenerative
Technology, Inc. as of December 31, 2023 and 2022 and for the years then ended, which report is included in this Annual Report on
Form 10-K of Harvard Apparatus Regenerative Technology, Inc. for the year ended December 31, 2023.
| /s/
Marcum LLP |
|
| |
|
| Marcum
LLP |
|
| Boston,
Massachusetts |
|
| March 28, 2024 |
|
EXHIBIT
31.1
Certification
I,
Junli He, certify that:
| 1. |
I
have reviewed this Annual Report on Form 10-K of Harvard Apparatus Regenerative Technology, Inc.; |
| |
|
| 2. |
Based
on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary
to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to
the period covered by this report; |
| |
|
| 3. |
Based
on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material
respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in
this report; |
| |
|
| 4. |
The
registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures
(as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)), and internal control over financial reporting (as defined in Exchange
Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| |
a. |
Designed
such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision,
to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others
within those entities, particularly during the period in which this report is being prepared; |
| |
|
|
| |
b. |
Designed
such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our
supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements
for external purposes in accordance with generally accepted accounting principles; |
| |
|
|
| |
c. |
Evaluated
the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about
the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation;
and |
| |
|
|
| |
d. |
Disclosed
in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s
most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected,
or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. |
The
registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over
financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or
persons performing the equivalent functions): |
| |
a. |
All
significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are
reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information;
and |
| |
|
|
| |
b. |
Any
fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s
internal control over financial reporting. |
Date:
March 28, 2024
| |
/s/
Junli (Jerry) He |
| |
Junli
(Jerry) He |
| |
Chief
Executive Officer, Director, and Chairman |
EXHIBIT
31.2
Certification
I,
Joseph Damasio Jr., certify that:
| 1. |
I
have reviewed this Annual Report on Form 10-K of Harvard Apparatus Regenerative Technology, Inc.; |
| |
|
| 2. |
Based
on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary
to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to
the period covered by this report; |
| |
|
| 3. |
Based
on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material
respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in
this report; |
| |
|
| 4. |
The
registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures
(as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)), and internal control over financial reporting (as defined in Exchange
Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| |
a. |
Designed
such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision,
to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others
within those entities, particularly during the period in which this report is being prepared; |
| |
|
|
| |
b. |
Designed
such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our
supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements
for external purposes in accordance with generally accepted accounting principles; |
| |
|
|
| |
c. |
Evaluated
the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about
the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation;
and |
| |
|
|
| |
d. |
Disclosed
in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s
most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected,
or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. |
The
registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over
financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or
persons performing the equivalent functions): |
| |
a. |
All
significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are
reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information;
and |
| |
|
|
| |
b. |
Any
fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s
internal control over financial reporting. |
Date:
March 28, 2024
| |
/s/
Joseph Damasio Jr. |
| |
Joseph
Damasio Jr. |
| |
Chief
Financial Officer |
EXHIBIT
32.1
CERTIFICATION
OF PERIODIC FINANCIAL REPORT
PURSUANT
TO 18 U.S.C. SECTION 1350
The
undersigned officer of Harvard Apparatus Regenerative Technology, Inc. (the Company) hereby certifies to his knowledge that the Company’s
Annual Report on Form 10-K for the year ended December 31, 2023 (the Report) to which this certification is being furnished as an exhibit,
as filed with the Securities and Exchange Commission on the date hereof, fully complies with the requirements of Section 13(a) or 15(d),
as applicable, of the Securities Exchange Act of 1934, as amended (the Exchange Act), and that the information contained in the Report
fairly presents, in all material respects, the financial condition and results of operations of the Company. This certification is provided
solely pursuant to 18 U.S.C. Section 1350 and Item 601(b)(32) of Regulation S-K (Item 601(b)(32)) promulgated under the Securities Act
of 1933, as amended (the Securities Act), and the Exchange Act. In accordance with clause (ii) of Item 601(b)(32), this certification
(A) shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of
that section, and (B) shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act,
except to the extent that the Company specifically incorporates it by reference.
Date:
March 28, 2024
| |
|
/s/
Junli (Jerry) He |
| |
Name: |
Junli
(Jerry) He |
| |
Title: |
Chief
Executive Officer, Director, and Chairman |
EXHIBIT
32.2
CERTIFICATION
OF PERIODIC FINANCIAL REPORT
PURSUANT
TO 18 U.S.C. SECTION 1350
The
undersigned officer of Harvard Apparatus Regenerative Technology, Inc. (the Company) hereby certifies to his knowledge that the Company’s
Annual Report on Form 10-K for the year ended December 31, 2023 (the Report) to which this certification is being furnished as an exhibit,
as filed with the Securities and Exchange Commission on the date hereof, fully complies with the requirements of Section 13(a) or 15(d),
as applicable, of the Securities Exchange Act of 1934, as amended (the Exchange Act), and that the information contained in the Report
fairly presents, in all material respects, the financial condition and results of operations of the Company. This certification is provided
solely pursuant to 18 U.S.C. Section 1350 and Item 601(b)(32) of Regulation S-K (Item 601(b)(32)) promulgated under the Securities Act
of 1933, as amended (the Securities Act), and the Exchange Act. In accordance with clause (ii) of Item 601(b)(32), this certification
(A) shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of
that section, and (B) shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act,
except to the extent that the Company specifically incorporates it by reference.
Date:
March 28, 2024
| |
|
/s/
Joseph Damasio Jr. |
| |
Name: |
Joseph
Damasio Jr. |
| |
Title: |
Chief
Financial Officer |
v3.24.1
Cover - USD ($)
$ in Millions |
12 Months Ended |
|
|
Dec. 31, 2023 |
Mar. 18, 2024 |
Jun. 30, 2023 |
| Cover [Abstract] |
|
|
|
| Document Type |
10-K
|
|
|
| Amendment Flag |
false
|
|
|
| Document Annual Report |
true
|
|
|
| Document Transition Report |
false
|
|
|
| Document Period End Date |
Dec. 31, 2023
|
|
|
| Document Fiscal Period Focus |
FY
|
|
|
| Document Fiscal Year Focus |
2023
|
|
|
| Current Fiscal Year End Date |
--12-31
|
|
|
| Entity File Number |
001-35853
|
|
|
| Entity Registrant Name |
Harvard
Apparatus Regenerative Technology, Inc.
|
|
|
| Entity Central Index Key |
0001563665
|
|
|
| Entity Tax Identification Number |
45-5210462
|
|
|
| Entity Incorporation, State or Country Code |
DE
|
|
|
| Entity Address, Address Line One |
84
October Hill Road
|
|
|
| Entity Address, Address Line Two |
Suite 11
|
|
|
| Entity Address, City or Town |
Holliston
|
|
|
| Entity Address, State or Province |
MA
|
|
|
| Entity Address, Postal Zip Code |
01746
|
|
|
| City Area Code |
(774)
|
|
|
| Local Phone Number |
233-7300
|
|
|
| Title of 12(g) Security |
Common
Stock, $0.01 par value
|
|
|
| Entity Well-known Seasoned Issuer |
No
|
|
|
| Entity Voluntary Filers |
No
|
|
|
| Entity Current Reporting Status |
Yes
|
|
|
| Entity Interactive Data Current |
Yes
|
|
|
| Entity Filer Category |
Non-accelerated Filer
|
|
|
| Entity Small Business |
true
|
|
|
| Entity Emerging Growth Company |
false
|
|
|
| Entity Shell Company |
false
|
|
|
| Entity Public Float |
|
|
$ 33.8
|
| Entity Common Stock, Shares Outstanding |
|
13,947,324
|
|
| Documents Incorporated by Reference [Text Block] |
Portions
of the Company’s definitive Proxy Statement in connection with the 2024 Annual Meeting of Stockholders, to be filed within 120
days after the end of the Registrant’s fiscal year, are incorporated by reference into Part III of this Form 10-K. Except with
respect to information specifically incorporated by reference in this Form 10-K, such Proxy Statement is not deemed to be filed as part
hereof.
|
|
|
| ICFR Auditor Attestation Flag |
false
|
|
|
| Document Financial Statement Error Correction [Flag] |
false
|
|
|
| Auditor Name |
Marcum
LLP
|
|
|
| Auditor Location |
Boston,
MA
|
|
|
| Auditor Firm ID |
688
|
|
|
| X |
- DefinitionBoolean flag that is true when the XBRL content amends previously-filed or accepted submission.
| Name: |
dei_AmendmentFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPCAOB issued Audit Firm Identifier Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorFirmId |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:nonemptySequenceNumberItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorLocation |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:internationalNameItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorName |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:internationalNameItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionEnd date of current fiscal year in the format --MM-DD.
| Name: |
dei_CurrentFiscalYearEndDate |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:gMonthDayItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true only for a form used as an annual report. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_DocumentAnnualReport |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicates whether any of the financial statement period in the filing include a restatement due to error correction. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Regulation S-K
-Number 229
-Section 402
-Subsection w
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 4: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_DocumentFinStmtErrorCorrectionFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFiscal period values are FY, Q1, Q2, and Q3. 1st, 2nd and 3rd quarter 10-Q or 10-QT statements have value Q1, Q2, and Q3 respectively, with 10-K, 10-KT or other fiscal year statements having FY.
| Name: |
dei_DocumentFiscalPeriodFocus |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:fiscalPeriodItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThis is focus fiscal year of the document report in YYYY format. For a 2006 annual report, which may also provide financial information from prior periods, fiscal 2006 should be given as the fiscal year focus. Example: 2006.
| Name: |
dei_DocumentFiscalYearFocus |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:gYearItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFor the EDGAR submission types of Form 8-K: the date of the report, the date of the earliest event reported; for the EDGAR submission types of Form N-1A: the filing date; for all other submission types: the end of the reporting or transition period. The format of the date is YYYY-MM-DD.
| Name: |
dei_DocumentPeriodEndDate |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:dateItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true only for a form used as a transition report. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Forms 10-K, 10-Q, 20-F
-Number 240
-Section 13
-Subsection a-1
| Name: |
dei_DocumentTransitionReport |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe type of document being provided (such as 10-K, 10-Q, 485BPOS, etc). The document type is limited to the same value as the supporting SEC submission type, or the word 'Other'.
| Name: |
dei_DocumentType |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:submissionTypeItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDocuments incorporated by reference. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-23
| Name: |
dei_DocumentsIncorporatedByReferenceTextBlock |
| Namespace Prefix: |
dei_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 1 such as Attn, Building Name, Street Name
| Name: |
dei_EntityAddressAddressLine1 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 2 such as Street or Suite number
| Name: |
dei_EntityAddressAddressLine2 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Definition
+ References
+ Details
| Name: |
dei_EntityAddressCityOrTown |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCode for the postal or zip code
| Name: |
dei_EntityAddressPostalZipCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionName of the state or province.
| Name: |
dei_EntityAddressStateOrProvince |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:stateOrProvinceItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionA unique 10-digit SEC-issued value to identify entities that have filed disclosures with the SEC. It is commonly abbreviated as CIK. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityCentralIndexKey |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:centralIndexKeyItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate number of shares or other units outstanding of each of registrant's classes of capital or common stock or other ownership interests, if and as stated on cover of related periodic report. Where multiple classes or units exist define each class/interest by adding class of stock items such as Common Class A [Member], Common Class B [Member] or Partnership Interest [Member] onto the Instrument [Domain] of the Entity Listings, Instrument.
| Name: |
dei_EntityCommonStockSharesOutstanding |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionIndicate 'Yes' or 'No' whether registrants (1) have filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that registrants were required to file such reports), and (2) have been subject to such filing requirements for the past 90 days. This information should be based on the registrant's current or most recent filing containing the related disclosure.
| Name: |
dei_EntityCurrentReportingStatus |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate if registrant meets the emerging growth company criteria. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityEmergingGrowthCompany |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCommission file number. The field allows up to 17 characters. The prefix may contain 1-3 digits, the sequence number may contain 1-8 digits, the optional suffix may contain 1-4 characters, and the fields are separated with a hyphen.
| Name: |
dei_EntityFileNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:fileNumberItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate whether the registrant is one of the following: Large Accelerated Filer, Accelerated Filer, Non-accelerated Filer. Definitions of these categories are stated in Rule 12b-2 of the Exchange Act. This information should be based on the registrant's current or most recent filing containing the related disclosure. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityFilerCategory |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:filerCategoryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTwo-character EDGAR code representing the state or country of incorporation.
| Name: |
dei_EntityIncorporationStateCountryCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:edgarStateCountryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Regulation S-T
-Number 232
-Section 405
| Name: |
dei_EntityInteractiveDataCurrent |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant's most recently completed second fiscal quarter.
| Name: |
dei_EntityPublicFloat |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionThe exact name of the entity filing the report as specified in its charter, which is required by forms filed with the SEC. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityRegistrantName |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the registrant is a shell company as defined in Rule 12b-2 of the Exchange Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityShellCompany |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicates that the company is a Smaller Reporting Company (SRC). Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntitySmallBusiness |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe Tax Identification Number (TIN), also known as an Employer Identification Number (EIN), is a unique 9-digit value assigned by the IRS. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityTaxIdentificationNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:employerIdItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate 'Yes' or 'No' if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
| Name: |
dei_EntityVoluntaryFilers |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate 'Yes' or 'No' if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Is used on Form Type: 10-K, 10-Q, 8-K, 20-F, 6-K, 10-K/A, 10-Q/A, 20-F/A, 6-K/A, N-CSR, N-Q, N-1A. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Securities Act
-Number 230
-Section 405
| Name: |
dei_EntityWellKnownSeasonedIssuer |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_IcfrAuditorAttestationFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLocal phone number for entity.
| Name: |
dei_LocalPhoneNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTitle of a 12(g) registered security. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection g
| Name: |
dei_Security12gTitle |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:securityTitleItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Consolidated Balance Sheets - USD ($)
$ in Thousands |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Current assets: |
|
|
| Cash and cash equivalents |
$ 432
|
$ 1,241
|
| Accounts receivable |
4
|
|
| Inventory |
50
|
|
| Prepaid research and development |
210
|
274
|
| Prepaid expenses and other current assets |
87
|
79
|
| Total current assets |
783
|
1,594
|
| Property, plant and equipment, net |
25
|
49
|
| Right-of-use assets, net |
48
|
147
|
| Deferred financing costs |
544
|
610
|
| Long-term prepaid contracts |
1,214
|
|
| Total assets |
2,614
|
2,400
|
| Current liabilities: |
|
|
| Accounts payable |
445
|
682
|
| Accrued and other current liabilities |
475
|
582
|
| Operating lease liability, current |
48
|
99
|
| Total current liabilities |
968
|
1,363
|
| Operating lease liability, net of current portion |
|
48
|
| Total liabilities |
968
|
1,411
|
| Commitments and contingencies (Note 9) |
|
|
| Series E convertible preferred stock, par value $0.01 per share, 5,000 shares authorized; 0 and 4,180 shares issued and outstanding at December 31, 2023 and 2022, respectively |
|
4,180
|
| Stockholders’ equity (deficit): |
|
|
| Common stock, par value $0.01 per share, 60,000,000 shares authorized; 13,947,324 and 12,174,467 issued and outstanding at December 31, 2023 and 2022, respectively |
139
|
122
|
| Additional paid-in capital |
93,463
|
79,698
|
| Accumulated deficit |
(91,956)
|
(83,011)
|
| Total stockholders’ equity (deficit) |
1,646
|
(3,191)
|
| Total liabilities and stockholders’ equity (deficit) |
$ 2,614
|
$ 2,400
|
| X |
- DefinitionDeferred financing cost non-current.
| Name: |
HRGN_DeferredFinancingCostNonCurrent |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionPrepaid research and development current.
| Name: |
HRGN_PrepaidResearchAndDevelopmentCurrent |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionCarrying value as of the balance sheet date of liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received that are used in an entity's business. Used to reflect the current portion of the liabilities (due within one year or within the normal operating cycle if longer). Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AccountsPayableCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount, after allowance for credit loss, of right to consideration from customer for product sold and service rendered in normal course of business, classified as current. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481990/310-10-45-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481990/310-10-45-9
| Name: |
us-gaap_AccountsReceivableNetCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of expenses incurred but not yet paid nor invoiced, and liabilities classified as other.
| Name: |
us-gaap_AccruedLiabilitiesAndOtherLiabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of excess of issue price over par or stated value of stock and from other transaction involving stock or stockholder. Includes, but is not limited to, additional paid-in capital (APIC) for common and preferred stock. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30)(a)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AdditionalPaidInCapital |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all assets that are recognized. Assets are probable future economic benefits obtained or controlled by an entity as a result of past transactions or events. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(12))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 23: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 26: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(11))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_Assets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all assets that are expected to be realized in cash, sold, or consumed within one year (or the normal operating cycle, if longer). Assets are probable future economic benefits obtained or controlled by an entity as a result of past transactions or events. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
| Name: |
us-gaap_AssetsCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_AssetsCurrentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of currency on hand as well as demand deposits with banks or financial institutions. Includes other kinds of accounts that have the general characteristics of demand deposits. Also includes short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Excludes cash and cash equivalents within disposal group and discontinued operation. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-4
| Name: |
us-gaap_CashAndCashEquivalentsAtCarryingValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionRepresents the caption on the face of the balance sheet to indicate that the entity has entered into (1) purchase or supply arrangements that will require expending a portion of its resources to meet the terms thereof, and (2) is exposed to potential losses or, less frequently, gains, arising from (a) possible claims against a company's resources due to future performance under contract terms, and (b) possible losses or likely gains from uncertainties that will ultimately be resolved when one or more future events that are deemed likely to occur do occur or fail to occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(15))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 942
-SubTopic 210
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03.17)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.25)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommitmentsAndContingencies |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAggregate par or stated value of issued nonredeemable common stock (or common stock redeemable solely at the option of the issuer). This item includes treasury stock repurchased by the entity. Note: elements for number of nonredeemable common shares, par value and other disclosure concepts are in another section within stockholders' equity. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount after valuation and LIFO reserves of inventory expected to be sold, or consumed within one year or operating cycle, if longer. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(6))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_InventoryNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all liabilities that are recognized. Liabilities are probable future sacrifices of economic benefits arising from present obligations of an entity to transfer assets or provide services to other entities in the future. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(14))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 21: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 22: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19-26)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_Liabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of liabilities and equity items, including the portion of equity attributable to noncontrolling interests, if any. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(32))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_LiabilitiesAndStockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionTotal obligations incurred as part of normal operations that are expected to be paid during the following twelve months or within one business cycle, if longer. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-5
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 21: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.21)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_LiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_LiabilitiesCurrentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as current. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as noncurrent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's right to use underlying asset under operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseRightOfUseAsset |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of asset related to consideration paid in advance for costs that provide economic benefits in future periods, and amount of other assets that are expected to be realized or consumed within one year or the normal operating cycle, if longer. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PrepaidExpenseAndOtherAssetsCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of amounts paid in advance for expenses which will be charged against earnings in periods after one year or beyond the operating cycle, if longer. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PrepaidExpenseNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount after accumulated depreciation, depletion and amortization of physical assets used in the normal conduct of business to produce goods and services and not intended for resale. Examples include, but are not limited to, land, buildings, machinery and equipment, office equipment, and furniture and fixtures. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 360
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480842/942-360-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of accumulated undistributed earnings (deficit). Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (g)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (h)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480990/946-20-50-11
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(23)(a)(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30)(a)(3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_RetainedEarningsAccumulatedDeficit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of equity (deficit) attributable to parent. Excludes temporary equity and equity attributable to noncontrolling interest. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(6))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 8: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 9: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 11: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(31))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 13: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 14: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 4.E)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480418/310-10-S99-2
| Name: |
us-gaap_StockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_StockholdersEquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCarrying amount, attributable to parent, of an entity's issued and outstanding stock which is not included within permanent equity. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Includes any type of security that is redeemable at a fixed or determinable price or on a fixed or determinable date or dates, is redeemable at the option of the holder, or has conditions for redemption which are not solely within the control of the issuer. Includes stock with a put option held by an ESOP and stock redeemable by a holder only in the event of a change in control of the issuer. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(23)(a)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.E.Q2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
| Name: |
us-gaap_TemporaryEquityCarryingAmountAttributableToParent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.24.1
Consolidated Balance Sheets (Parenthetical) - $ / shares
|
Dec. 31, 2023 |
Dec. 31, 2022 |
| Statement of Financial Position [Abstract] |
|
|
| Series E convertible preferred stock, par value, per share |
$ 0.01
|
$ 0.01
|
| Series E convertible preferred stock, shares authorized |
5,000
|
5,000
|
| Series E convertible preferred stock, shares issued |
0
|
4,180
|
| Series E convertible preferred stock, shares outstanding |
0
|
4,180
|
| Common stock, par value (in dollars per share) |
$ 0.01
|
$ 0.01
|
| Common stock, shares authorized |
60,000,000
|
60,000,000
|
| Common stock, shares issued |
13,947,324
|
12,174,467
|
| Common stock, shares outstanding |
13,947,324
|
12,174,467
|
| X |
- DefinitionFace amount or stated value per share of common stock. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockParOrStatedValuePerShare |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe maximum number of common shares permitted to be issued by an entity's charter and bylaws. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionTotal number of common shares of an entity that have been sold or granted to shareholders (includes common shares that were issued, repurchased and remain in the treasury). These shares represent capital invested by the firm's shareholders and owners, and may be all or only a portion of the number of shares authorized. Shares issued include shares outstanding and shares held in the treasury. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesIssued |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares of common stock outstanding. Common stock represent the ownership interest in a corporation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_StatementOfFinancialPositionAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPer share amount of par value or stated value of stock classified as temporary equity. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 480
-SubTopic 10
-Section S99
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480244/480-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (27)
-SubTopic 10
-Topic 210
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_TemporaryEquityParOrStatedValuePerShare |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe maximum number of securities classified as temporary equity that are permitted to be issued by an entity's charter and bylaws. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Includes any type of security that is redeemable at a fixed or determinable price or on a fixed or determinable date or dates, is redeemable at the option of the holder, or has conditions for redemption which are not solely within the control of the issuer. If convertible, the issuer does not control the actions or events necessary to issue the maximum number of shares that could be required to be delivered under the conversion option if the holder exercises the option to convert the stock to another class of equity. If the security is a warrant or a rights issue, the warrant or rights issue is considered to be temporary equity if the issuer cannot demonstrate that it would be able to deliver upon the exercise of the option by the holder in all cases. Includes stock with put option held by ESOP and stock redeemable by holder only in the event of a change in control of the issuer. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(27)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_TemporaryEquitySharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe number of securities classified as temporary equity that have been sold (or granted) to the entity's shareholders. Securities issued include securities outstanding and securities held in treasury. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Includes any type of security that is redeemable at a fixed or determinable price or on a fixed or determinable date or dates, is redeemable at the option of the holder, or has conditions for redemption which are not solely within the control of the issuer. If convertible, the issuer does not control the actions or events necessary to issue the maximum number of shares that could be required to be delivered under the conversion option if the holder exercises the option to convert the stock to another class of equity. If the security is a warrant or a rights issue, the warrant or rights issue is considered to be temporary equity if the issuer cannot demonstrate that it would be able to deliver upon the exercise of the option by the holder in all cases. Includes stock with put option held by ESOP and stock redeemable by holder only in the event of a change in control of the issuer. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(27)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_TemporaryEquitySharesIssued |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe number of securities classified as temporary equity that have been issued and are held by the entity's shareholders. Securities outstanding equals securities issued minus securities held in treasury. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Includes any type of security that is redeemable at a fixed or determinable price or on a fixed or determinable date or dates, is redeemable at the option of the holder, or has conditions for redemption which are not solely within the control of the issuer. If convertible, the issuer does not control the actions or events necessary to issue the maximum number of shares that could be required to be delivered under the conversion option if the holder exercises the option to convert the stock to another class of equity. If the security is a warrant or a rights issue, the warrant or rights issue is considered to be temporary equity if the issuer cannot demonstrate that it would be able to deliver upon the exercise of the option by the holder in all cases. Includes stock with put option held by ESOP and stock redeemable by holder only in the event of a change in control of the issuer. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(27)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_TemporaryEquitySharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
v3.24.1
Consolidated Statements of Operations - USD ($)
$ in Thousands |
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Income Statement [Abstract] |
|
|
| Product revenue |
$ 103
|
|
| Operating expenses: |
|
|
| Cost of sales |
24
|
|
| Research and development |
3,062
|
1,742
|
| Sales and marketing |
294
|
|
| General and administrative |
5,713
|
4,411
|
| Total operating expenses |
9,093
|
6,153
|
| Operating loss |
(8,990)
|
(6,153)
|
| Other income (expense), net: |
|
|
| Sublease income |
|
87
|
| Change in fair value of warrant liability |
|
2
|
| Interest income |
64
|
|
| Interest expense |
(14)
|
(9)
|
| Other expense |
(5)
|
|
| Total other income, net |
45
|
80
|
| Net loss |
(8,945)
|
(6,073)
|
| Preferred stock dividends |
(77)
|
(180)
|
| Net loss attributable to common stockholders |
$ (9,022)
|
$ (6,253)
|
| Basic net loss per share attributable to common stockholders |
$ (0.67)
|
$ (0.55)
|
| Diluted net loss per share attributable to common stockholders |
$ (0.67)
|
$ (0.55)
|
| Weighted average common shares, basic |
13,455,666
|
11,349,610
|
| Weighted average common shares, diluted |
13,455,666
|
11,349,610
|
| X |
- Definition
+ References
+ Details
| Name: |
HRGN_SubleaseExpenseIncome |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period per each share of common stock or unit outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period available to each share of common stock or common unit outstanding during the reporting period and to each share or unit that would have been outstanding assuming the issuance of common shares or units for all dilutive potential common shares or units outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareDiluted |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of expense (income) related to adjustment to fair value of warrant liability. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 25
-Paragraph 13
-SubTopic 10
-Topic 480
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481766/480-10-25-13
| Name: |
us-gaap_FairValueAdjustmentOfWarrants |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate total of expenses of managing and administering the affairs of an entity, including affiliates of the reporting entity, which are not directly or indirectly associated with the manufacture, sale or creation of a product or product line. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(2)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03.4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
| Name: |
us-gaap_GeneralAndAdministrativeExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_IncomeStatementAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of interest income and other income recognized during the period. Included in this element is interest derived from investments in debt securities, cash and cash equivalents, and other investments which reflect the time value of money or transactions in which the payments are for the use or forbearance of money and other income from ancillary business-related activities (that is, excluding major activities considered part of the normal operations of the business).
| Name: |
us-gaap_InterestAndOtherIncome |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of the cost of borrowed funds accounted for as interest expense. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 3
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 835
-SubTopic 30
-Section 45
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482925/835-30-45-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04.9)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (210.5-03(11))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 835
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483013/835-20-50-1
| Name: |
us-gaap_InterestExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, after deduction of tax, noncontrolling interests, dividends on preferred stock and participating securities; of income (loss) available to common shareholders. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 5
-Subparagraph (SAB Topic 6.B)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-5
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
Reference 11: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-11
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
| Name: |
us-gaap_NetIncomeLossAvailableToCommonStockholdersBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate amount of income or expense from ancillary business-related activities (that is to say, excluding major activities considered part of the normal operations of the business). Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03.7)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
| Name: |
us-gaap_NonoperatingIncomeExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionGenerally recurring costs associated with normal operations except for the portion of these expenses which can be clearly related to production and included in cost of sales or services. Includes selling, general and administrative expense.
| Name: |
us-gaap_OperatingExpenses |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_OperatingExpensesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe net result for the period of deducting operating expenses from operating revenues. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 4: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
| Name: |
us-gaap_OperatingIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of income (expense) related to nonoperating activities, classified as other. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03.9)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
| Name: |
us-gaap_OtherNonoperatingIncomeExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_OtherNonoperatingIncomeExpenseAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate costs incurred (1) in a planned search or critical investigation aimed at discovery of new knowledge with the hope that such knowledge will be useful in developing a new product or service, a new process or technique, or in bringing about a significant improvement to an existing product or process; or (2) to translate research findings or other knowledge into a plan or design for a new product or process or for a significant improvement to an existing product or process whether intended for sale or the entity's use, during the reporting period charged to research and development projects, including the costs of developing computer software up to the point in time of achieving technological feasibility, and costs allocated in accounting for a business combination to in-process projects deemed to have no alternative future use. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 730
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482916/730-10-50-1
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 912
-SubTopic 730
-Name Accounting Standards Codification
-Section 25
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482517/912-730-25-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 985
-SubTopic 20
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481283/985-20-50-1
| Name: |
us-gaap_ResearchAndDevelopmentExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, excluding tax collected from customer, of revenue from satisfaction of performance obligation by transferring promised good or service to customer. Tax collected from customer is tax assessed by governmental authority that is both imposed on and concurrent with specific revenue-producing transaction, including, but not limited to, sales, use, value added and excise. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 924
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 11.L)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479941/924-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 42
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-42
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 40
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-40
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-4
| Name: |
us-gaap_RevenueFromContractWithCustomerExcludingAssessedTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate total amount of expenses directly related to the marketing or selling of products or services.
| Name: |
us-gaap_SellingAndMarketingExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAccretion of temporary equity during the period due to cash, stock, and in-kind dividends. This item is an adjustment to net income necessary to derive net income apportioned to common stockholders and is to be distinguished from Temporary Equity, Accretion of Dividends (Temporary Equity, Accretion of Dividends).
| Name: |
us-gaap_TemporaryEquityDividendsAdjustment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe average number of shares or units issued and outstanding that are used in calculating diluted EPS or earnings per unit (EPU), determined based on the timing of issuance of shares or units in the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 16
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-16
| Name: |
us-gaap_WeightedAverageNumberOfDilutedSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of [basic] shares or units, after adjustment for contingently issuable shares or units and other shares or units not deemed outstanding, determined by relating the portion of time within a reporting period that common shares or units have been outstanding to the total time in that period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
| Name: |
us-gaap_WeightedAverageNumberOfSharesOutstandingBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Consolidated Statements of Changes in Stockholders' Equity (Deficit) - USD ($)
$ in Thousands |
Preferred Stock [Member]
Series E Convertible Preferred Stock [Member]
|
Common Stock [Member] |
Additional Paid-in Capital [Member] |
Retained Earnings [Member] |
Total |
| Beginning balance, value at Dec. 31, 2021 |
|
$ 108
|
$ 73,801
|
$ (76,938)
|
$ (3,029)
|
| Temporary equity, beginning balance at Dec. 31, 2021 |
|
|
|
|
|
| Beginning Balance, shares at Dec. 31, 2021 |
|
10,760,871
|
|
|
|
| Net loss |
|
|
|
(6,073)
|
(6,073)
|
| Share-based compensation |
|
|
1,031
|
|
1,031
|
| Issuance of series E convertible preferred stock |
4,000
|
|
|
|
|
| Issuance of series E convertible preferred stock |
4,000
|
|
|
|
|
| Preferred stock dividends |
180
|
|
(180)
|
|
(180)
|
| Temporary equity, preferred stock dividends |
180
|
|
|
|
|
| Issuance of common stock and warrants to purchase common stock |
|
$ 8
|
5,052
|
|
5,060
|
| Issuance of common stock and warrants to purchase common stock (in shares) |
|
854,771
|
|
|
|
| Issuance of common stock from exercise of warrants |
|
$ 6
|
(6)
|
|
|
| Issuance of common stock from exercise of warrants (in shares) |
|
558,825
|
|
|
|
| Ending balance, value at Dec. 31, 2022 |
4,180
|
$ 122
|
79,698
|
(83,011)
|
(3,191)
|
| Temporary equity, ending balance at Dec. 31, 2022 |
4,180
|
|
|
|
4,180
|
| Ending Balance, shares at Dec. 31, 2022 |
|
12,174,467
|
|
|
|
| Net loss |
|
|
|
(8,945)
|
(8,945)
|
| Share-based compensation |
|
|
3,461
|
|
3,461
|
| Preferred stock dividends |
77
|
|
(77)
|
|
(77)
|
| Temporary equity, preferred stock dividends |
77
|
|
|
|
|
| Conversion of preferred stock for common stock |
(4,257)
|
$ 7
|
4,250
|
|
4,257
|
| Temporary equity, conversion of preferred stock for common stock |
(4,257)
|
|
|
|
|
| Conversion of preferred stock for common stock, shares |
|
706,626
|
|
|
|
| Issuance of common stock |
|
$ 10
|
5,982
|
|
5,992
|
| Issuance of common stock, shares |
|
1,000,967
|
|
|
|
| Issuance of common stock from exercise of options |
|
|
149
|
|
149
|
| Issuance of common stock, net of offering costs, shares |
|
65,264
|
|
|
|
| Ending balance, value at Dec. 31, 2023 |
|
$ 139
|
$ 93,463
|
$ (91,956)
|
1,646
|
| Temporary equity, ending balance at Dec. 31, 2023 |
|
|
|
|
|
| Ending Balance, shares at Dec. 31, 2023 |
|
13,947,324
|
|
|
|
| X |
- DefinitionIssuance of common stock and warrants to purchase common stock share net of offering costs.
| Name: |
HRGN_IssuanceOfCommonStockAndWarrantsToPurchaseCommonStockShareNetOfOfferingCosts |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIssuance of common stock and warrants to purchase common stock value net of offering costs.
| Name: |
HRGN_IssuanceOfCommonStockAndWarrantsToPurchaseCommonStockValueNetOfOfferingCosts |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionIssuance of common stock from exercise of warrants.
| Name: |
HRGN_IssuanceOfCommonStockFromExerciseOfWarrants |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period shares issuance of common stock from exercise of options.
| Name: |
HRGN_StockIssuedDuringPeriodSharesIssuanceOfCommonStockFromExerciseOfOptions |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value convertible preferred stock.
| Name: |
HRGN_StockIssuedDuringPeriodValueConvertiblePreferredStock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value issuance of common stock from exercise of options.
| Name: |
HRGN_StockIssuedDuringPeriodValueIssuanceOfCommonStockFromExerciseOfOptions |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value of common stock upon warrant exercises.
| Name: |
HRGN_StockIssuedDuringPeriodValueOfCommonStockUponWarrantExercises |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of decrease in additional paid in capital (APIC) resulting from dividends legally declared (or paid) in excess of retained earnings balance. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
| Name: |
us-gaap_AdjustmentsToAdditionalPaidInCapitalDividendsInExcessOfRetainedEarnings |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase to additional paid-in capital (APIC) for recognition of cost for award under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 20
-Section 55
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481089/718-20-55-13
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 20
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481089/718-20-55-12
| Name: |
us-gaap_AdjustmentsToAdditionalPaidInCapitalSharebasedCompensationRequisiteServicePeriodRecognitionValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares issued which are neither cancelled nor held in the treasury.
| Name: |
us-gaap_SharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares issued during the period as a result of the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1E
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481139/470-20-50-1E
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-30)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of new stock issued during the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(i)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe gross value of stock issued during the period upon the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-31)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionEquity impact of the value of new stock issued during the period. Includes shares issued in an initial public offering or a secondary public offering. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-11
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 205
-Name Accounting Standards Codification
-Section 45
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480767/946-205-45-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of equity (deficit) attributable to parent. Excludes temporary equity and equity attributable to noncontrolling interest. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(6))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 8: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 9: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 11: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(31))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 13: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 14: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 4.E)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480418/310-10-S99-2
| Name: |
us-gaap_StockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionValue of accretion of temporary equity during the period due to unpaid dividends.
| Name: |
us-gaap_TemporaryEquityAccretionOfDividends |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionCarrying amount, attributable to parent, of an entity's issued and outstanding stock which is not included within permanent equity. Temporary equity is a security with redemption features that are outside the control of the issuer, is not classified as an asset or liability in conformity with GAAP, and is not mandatorily redeemable. Includes any type of security that is redeemable at a fixed or determinable price or on a fixed or determinable date or dates, is redeemable at the option of the holder, or has conditions for redemption which are not solely within the control of the issuer. Includes stock with a put option held by an ESOP and stock redeemable by a holder only in the event of a change in control of the issuer. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(23)(a)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.E.Q2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
| Name: |
us-gaap_TemporaryEquityCarryingAmountAttributableToParent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of increase (decrease) in temporary equity from changes classified as other.
| Name: |
us-gaap_TemporaryEquityOtherChanges |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionValue of new stock classified as temporary equity issued during the period.
| Name: |
us-gaap_TemporaryEquityStockIssuedDuringPeriodValueNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
v3.24.1
Consolidated Statements of Cash Flows - USD ($)
|
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| OPERATING ACTIVITIES |
|
|
| Net loss |
$ (8,945,000)
|
$ (6,073,000)
|
| Adjustments to reconcile net loss to net cash and cash equivalents used in operating activities: |
|
|
| Share-based compensation expense |
3,461,000
|
1,031,000
|
| Depreciation |
35,000
|
52,000
|
| Amortization of operating right-of-use assets |
99,000
|
22,000
|
| Change in fair value of warrant liability |
|
(2,000)
|
| Changes in operating assets and liabilities: |
|
|
| Accounts receivable |
(4,000)
|
|
| Inventory |
(50,000)
|
|
| Prepaid research and development |
64,000
|
(274,000)
|
| Prepaid expenses and other current assets |
(8,000)
|
216,000
|
| Deferred financing costs |
66,000
|
(610,000)
|
| Long-term prepaid contracts |
(1,214,000)
|
|
| Accounts payable |
(237,000)
|
6,000
|
| Operating lease liability |
(99,000)
|
(22,000)
|
| Accrued and other current liabilities |
(107,000)
|
548,000
|
| Net cash used in operating activities |
(6,939,000)
|
(5,106,000)
|
| INVESTING ACTIVITIES |
|
|
| Purchases of short-term investments |
(2,523,000)
|
|
| Redemption of short-term investments |
2,523,000
|
|
| Purchases of property, plant and equipment |
(11,000)
|
(5,000)
|
| Net cash used in investing activities |
(11,000)
|
(5,000)
|
| FINANCING ACTIVITIES |
|
|
| Proceeds from issuance of common stock |
5,992,000
|
5,060,000
|
| Proceeds from exercise of stock options |
149,000
|
|
| Net cash provided by financing activities |
6,141,000
|
5,060,000
|
| Net decrease in cash and cash equivalents |
(809,000)
|
(51,000)
|
| Cash and cash equivalents at the beginning of the year |
1,241,000
|
1,292,000
|
| Cash and cash equivalents at the end of the year |
432,000
|
1,241,000
|
| SUPPLEMENTAL INFORMATION |
|
|
| Interest paid in cash |
14,000
|
9,000
|
| Supplemental disclosure of non-cash activities: |
|
|
| Settlement of contingency matter |
|
(3,250,000)
|
| Settlement of due to Harvard Bioscience included in accrued and other current liabilities |
|
(750,000)
|
| Issuance of Series E convertible preferred stock |
|
4,000,000
|
| Preferred stock dividends |
77,000
|
180,000
|
| Increase of right-of-use asset and liability due to lease extension |
|
$ 63,000
|
| X |
- DefinitionIncrease decrease in deferred financing costs.
| Name: |
HRGN_IncreaseDecreaseInDeferredFinancingCosts |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionIncrease decrease in prepaid research and development.
| Name: |
HRGN_IncreaseDecreaseInPrepaidResearchAndDevelopment |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionIncrease of right of use asset and liability due to lease extension.
| Name: |
HRGN_IncreaseOfRightOfUseAssetAndLiabilityDueToLeaseExtension |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of settlement to parent company.
| Name: |
HRGN_SettlementAmountDueToParentIncludedInAccruedAndOtherCurrentLiabilities |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of settlement of contingency in noncash activity.
| Name: |
HRGN_SettlementOfContingencyMatter |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_AdjustmentsToReconcileNetIncomeLossToCashProvidedByUsedInOperatingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash and cash equivalents, and cash and cash equivalents restricted to withdrawal or usage. Excludes amount for disposal group and discontinued operations. Cash includes, but is not limited to, currency on hand, demand deposits with banks or financial institutions, and other accounts with general characteristics of demand deposits. Cash equivalents include, but are not limited to, short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-8
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-4
| Name: |
us-gaap_CashCashEquivalentsRestrictedCashAndRestrictedCashEquivalents |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of increase (decrease) in cash, cash equivalents, and cash and cash equivalents restricted to withdrawal or usage; including effect from exchange rate change. Cash includes, but is not limited to, currency on hand, demand deposits with banks or financial institutions, and other accounts with general characteristics of demand deposits. Cash equivalents include, but are not limited to, short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-SubTopic 230
-Topic 830
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481877/830-230-45-1
| Name: |
us-gaap_CashCashEquivalentsRestrictedCashAndRestrictedCashEquivalentsPeriodIncreaseDecreaseIncludingExchangeRateEffect |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_CashFlowNoncashInvestingAndFinancingActivitiesDisclosureAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of expense recognized in the current period that reflects the allocation of the cost of tangible assets over the assets' useful lives. Includes production and non-production related depreciation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 360
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_Depreciation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of paid and unpaid preferred stock dividends declared with the form of settlement in cash, stock and payment-in-kind (PIK). Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-SubTopic 405
-Topic 942
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481071/942-405-45-2
| Name: |
us-gaap_DividendsPreferredStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of expense (income) related to adjustment to fair value of warrant liability. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 25
-Paragraph 13
-SubTopic 10
-Topic 480
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481766/480-10-25-13
| Name: |
us-gaap_FairValueAdjustmentOfWarrants |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in the aggregate amount of liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received that are used in an entity's business. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInAccountsPayable |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in amount due within one year (or one business cycle) from customers for the credit sale of goods and services. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInAccountsReceivable |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in accrued expenses, and obligations classified as other. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInAccruedLiabilitiesAndOtherOperatingLiabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in the aggregate value of all inventory held by the reporting entity, associated with underlying transactions that are classified as operating activities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInInventories |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_IncreaseDecreaseInOperatingCapitalAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in obligation for operating lease. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(1)
-SubTopic 20
-Topic 842
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_IncreaseDecreaseInOperatingLeaseLiability |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in prepaid expenses, and assets classified as other. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInPrepaidDeferredExpenseAndOtherAssets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in the amount of outstanding money paid in advance for goods or services that bring economic benefits for future periods. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInPrepaidExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash paid for interest, excluding capitalized interest, classified as operating activity. Includes, but is not limited to, payment to settle zero-coupon bond for accreted interest of debt discount and debt instrument with insignificant coupon interest rate in relation to effective interest rate of borrowing attributable to accreted interest of debt discount. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (e)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-2
| Name: |
us-gaap_InterestPaidNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from financing activities, including discontinued operations. Financing activity cash flows include obtaining resources from owners and providing them with a return on, and a return of, their investment; borrowing money and repaying amounts borrowed, or settling the obligation; and obtaining and paying for other resources obtained from creditors on long-term credit. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
| Name: |
us-gaap_NetCashProvidedByUsedInFinancingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInFinancingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from investing activities, including discontinued operations. Investing activity cash flows include making and collecting loans and acquiring and disposing of debt or equity instruments and property, plant, and equipment and other productive assets. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
| Name: |
us-gaap_NetCashProvidedByUsedInInvestingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInInvestingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from operating activities, including discontinued operations. Operating activity cash flows include transactions, adjustments, and changes in value not defined as investing or financing activities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-25
| Name: |
us-gaap_NetCashProvidedByUsedInOperatingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInOperatingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of periodic reduction over lease term of carrying amount of right-of-use asset from operating lease. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_OperatingLeaseRightOfUseAssetAmortizationExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash outflow associated with the purchase of all investments (debt, security, other) during the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-13
| Name: |
us-gaap_PaymentsToAcquireInvestments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash outflow associated with the acquisition of long-lived, physical assets that are used in the normal conduct of business to produce goods and services and not intended for resale; includes cash outflows to pay for construction of self-constructed assets. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 13
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-13
| Name: |
us-gaap_PaymentsToAcquirePropertyPlantAndEquipment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from the additional capital contribution to the entity. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfCommonStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from sales of all investments, including securities and other assets, having ready marketability and intended by management to be liquidated, if necessary, within the current operating cycle. Includes cash flows from securities classified as trading securities that were acquired for reasons other than sale in the short-term. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-12
| Name: |
us-gaap_ProceedsFromSaleOfShortTermInvestments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow from exercise of option under share-based payment arrangement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2A
-Subparagraph (a)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2A
| Name: |
us-gaap_ProceedsFromStockOptionsExercised |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of noncash expense for share-based payment arrangement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_ShareBasedCompensation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe fair value of stock issued in noncash financing activities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-4
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-3
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-5
| Name: |
us-gaap_StockIssued1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
v3.24.1
Organization
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Organization, Consolidation and Presentation of Financial Statements [Abstract] |
|
| Organization |
1.
Organization
Overview
Harvard
Apparatus Regenerative Technology, Inc. (Harvard Apparatus Regenerative Technology or the Company) is a biotechnology company with a
mission to cure patients of cancers, injuries, and birth defects of the gastro-intestinal tract and the airways. The Company believes
its technology is likely to be used to treat esophageal cancer, esophageal injuries, and birth defects in the esophagus. The Company
believes additional product candidates in its pipeline may treat intestinal cancer and colon cancer. Since inception, the Company has
devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, and
acquiring operating assets.
On
October 31, 2013, Harvard Bioscience, Inc., or Harvard Bioscience, contributed its regenerative medicine business assets, plus $15 million
of cash, into Harvard Apparatus Regenerative Technology, or the Separation. On November 1, 2013, the spin-off of the Company from Harvard
Bioscience was completed. On that date, the Company became an independent company that operates the regenerative medicine business previously
owned by Harvard Bioscience. The spin-off was completed through the distribution to Harvard Bioscience stockholders of all the shares
of common stock of Harvard Apparatus Regenerative Technology, or the Distribution.
Basis
of Presentation
The
consolidated financial statements reflect the Company’s financial position, results of operations and cash flows in conformity
with generally accepted accounting principles in the United States, or U.S. GAAP.
Going
Concern
The
Company has incurred substantial operating losses since its inception, and as of December 31, 2023 had an accumulated deficit of
approximately $92.0
million and will require additional financing to fund future operations. The Company expects that its operating cash on-hand as of
December 31, 2023 of approximately $0.4
million and debt financing of $0.5
million in gross proceeds received subsequent to December 31, 2023 will enable it to fund its operating expenses and capital
expenditure requirements only into the second quarter of 2024. Therefore, these conditions raise substantial doubt about the
Company’s ability to continue as a going concern.
The
Company will need to raise additional funds to fund its operations. In the event the Company does not raise additional capital from outside
sources during the first quarter of 2024, it may be forced to curtail or cease its operations. Cash requirements and cash resource
needs will vary significantly depending upon the timing of the financial and other resource needs that will be required to complete ongoing
development, pre-clinical and clinical testing of product candidates, as well as regulatory efforts and collaborative arrangements necessary
for the Company’s product candidates that are currently under development. The Company is currently seeking and will continue to
seek financings from other existing and/or new investors to raise necessary funds through a combination of public or private equity offerings.
The Company may also pursue debt financings, other financing mechanisms, research grants, or strategic collaborations and licensing arrangements.
The Company may not be able to obtain additional financing on favorable terms, if at all.
The
Company’s operations will be adversely affected if it is unable to raise or obtain needed funding and may materially affect the
Company’s ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming
that the Company will continue as a going concern and therefore, the consolidated financial statements do not include any adjustments
to reflect the possible future effects on the recoverability and classification of assets or the amount and classifications of liabilities
that may result from the outcome of this uncertainty.
|
| X |
- References
+ Details
| Name: |
us-gaap_OrganizationConsolidationAndPresentationOfFinancialStatementsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for organization, consolidation and basis of presentation of financial statements disclosure. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480424/946-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480424/946-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 810
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//810/tableOfContent
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 205
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//205/tableOfContent
| Name: |
us-gaap_OrganizationConsolidationAndPresentationOfFinancialStatementsDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Summary of Significant Accounting Policies
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Summary of Significant Accounting Policies |
2.
Summary of Significant Accounting Policies
Principles
of Consolidation
The
consolidated financial statements include the accounts of Harvard Apparatus Regenerative Technology, Inc. (Regenerative Biotech) and its three
wholly-owned subsidiaries, Harvard Apparatus Regenerative Technology Limited (Hong Kong), Harvard Apparatus Regenerative Technology
(Hangzhou) Limited (China) and Harvard Apparatus Regenerative Technology GmbH (Germany). All intercompany balances and transactions
have been eliminated in consolidation.
Use
of Estimates
The
process of preparing consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Such estimates include, but are not
limited to, share-based compensation, valuation of warrant liability, accrued expenses and the valuation allowance for deferred income
taxes. Actual results could differ from those estimates.
Revenue
We
recognize revenue in accordance with FASB ASC 606, Revenue from Contracts with Customers. We offer consumer products primarily
through a third-party online store. Revenue is recognized at a point in time when control of the goods is transferred to the customer,
which generally occurs upon the delivery to the customer. For any company direct sales to customers, revenue is recognized at a point
in time upon shipment of product or hand-delivery to customer. Revenue also excludes any amounts collected on behalf of third parties,
including sales and indirect taxes.
We
identify a performance obligation as distinct if both the following criteria are true: the customer can benefit from the good or service
either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer
the good or service to the customer is separately identifiable from other promises in the contract. Determining the standalone selling
price (“SSP”) and allocation of consideration from a contract to the individual performance obligations, and the appropriate
timing of revenue recognition, is the result of significant qualitative and quantitative judgments. Management considers a variety of
factors such as historical sales, usage rates, costs, and expected margin, which may vary over time depending upon the unique facts and
circumstances related to each performance obligation in making these estimates. While changes in the allocation of the SSP between performance
obligations will not affect the amount of total revenue recognized for a particular contract, any material changes could impact the timing
of revenue recognition, which would have a material effect on our financial position and result of operations. This is because the contract
consideration is allocated to each performance obligation, delivered or undelivered, at the inception of the contract based on the SSP
of each distinct performance obligation.
Cost
of Sales
Cost
of sales primarily consists of the purchase price of consumer products, taxes, inbound and outbound shipping costs. Shipping costs to
receive products from our suppliers are recognized as cost of sales when incurred. E-commerce processing and related transaction costs,
including those associated with seller transactions, are classified in sales and marketing on our consolidated statements of
operations.
Research
and Development
Research
and development costs are expensed as incurred.
Sales
and Marketing
Sales
and marketing costs include advertising and payroll and related expenses for personnel engaged in marketing and selling activities.
General
and Administrative
General
and administrative expenses primarily consist of costs for corporate functions, including payroll and related expenses; facilities and
equipment expenses, such as depreciation and amortization expense and rent; and professional fees.
Segment Information
The
Company manages its operations as two separate operating segments for the purposes of assessing performance and making operating
decisions. The Company has one operating unit focused on the development and commercialization of therapies to cure patients of
cancers, injuries, and birth defects of the gastro-intestinal tract and the airways. The other operating unit is focused on personal
healthcare through longevity dietary supplements. We have determined that our chief executive officer is the chief operating
decision maker (CODM). The CODM reviews financial information presented by operating unit. Resource allocation decisions are
made by the CODM based on operating unit results.
Cash
and Cash Equivalents
The
Company considers all highly liquid investments with a maturity of three months or less at the date of purchase to be cash
equivalents. The Company currently invests available cash in money market funds. As of December 31, 2023, the Company had
approximately $111,000
of cash equivalents in a money market fund.
Accounts Receivable
Allowances
are provided for estimated amounts of accounts receivable which may not be collected. At
December 31, 2023,
we determined that no allowance against accounts receivable was necessary.
Inventory
Inventory,
consisting of products available for sale, are primarily accounted for using the first-in, first-out method, and are valued at the lower
of cost or net realizable value.
We
maintain ownership of our inventory at the third-party warehouse, regardless of whether fulfillment is provided by us or the third-party
e-commerce seller, and therefore these products are included in our inventories.
Deferred Financing Costs
We capitalized costs relating to a
registered offering that we postponed in 2023 but expect to resume in the near future. The costs include payments made to attorneys,
accountants, regulators and consultants. Once we complete the registered offering, the deferred financing costs will be reclassified
to stockholders’ equity (deficit) on the consolidated balance sheets to offset the proceeds from the registered offering.
Long-term
prepaid contracts
We
have contracted with partners relating to our clinical trial activities. Upon execution of the contracts, we made initial payments
of $1.2
million as deposits recorded as long-term assets and will be applied against final invoices which are more than a year away. The deposits will be recorded as expense when the clinical trial is substantially
completed. Costs for the clinical trial activities throughout our clinical trial under these contracts are recognized as expense and payable
based on costs incurred.
Property,
Plant and Equipment
Property,
plant and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets
as follows:
Schedule of Property Plant and Equipment Estimated Useful Lives
| Leasehold improvements | |
Shorter of expected useful life
or lease term |
| Computer equipment and software | |
3 years |
| Furniture, machinery and equipment | |
5-7 years |
Maintenance
and repairs are charged to expense as incurred, while any additions or improvements are capitalized.
Impairment
of Long-Lived Assets
Assessments
of long-lived assets and the remaining useful lives of such long-lived assets are reviewed for impairment whenever a triggering event
occurs or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. An asset, or group of assets,
are considered to be impaired when the undiscounted estimated net cash flows expected to be generated by the asset, or group of assets,
are less than its carrying amount. The impairment recognized is the amount by which the carrying amount exceeds the fair market value
of the impaired asset, or group of assets, based on the present value of the expected future cash flows associated with the use of the
asset. Through December 31, 2023, no such impairment charges have been recorded.
Share-based
Compensation
The
Company measures all stock options and restricted stock awards granted to employees, directors and non-employees based on the fair value
on the date of the grant and recognizes compensation expense of those awards, net of forfeitures, over the requisite vesting period,
which is generally the service period of the respective award. Generally, the Company issues stock options and restricted stock awards
with only service-based vesting conditions on a straight-line basis over the requisite service period for the entire award (that is,
over the requisite service period of the last separately vesting portion of the award). Expense on share-based awards for which vesting
is performance or milestone based is recognized on a straight-line basis from the date when it is determined that the achievement of
the milestone is probable to the vesting/milestone achievement date.
The
Company elected to use the Black-Scholes option-pricing model for the valuation of stock-based payment awards. The determination of the
fair value of stock-based payment awards is determined on the date of grant using the Black-Scholes option-pricing model which is affected
by the market price as well as assumptions regarding a number of subjective variables. These variables include, but are not limited to,
its expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. When
performance-based grants are issued, the Company recognizes no expense until achievement of the performance requirement is deemed probable.
Share-based
compensation expense is based on awards ultimately expected to vest and has been reduced for annualized estimated forfeiture where the
minimum amount of expense recorded is at least equal to the percent of an award vested. Forfeitures are estimated based on historical
experience and weighting of various employee classes under the respective plan at the time of grant and revised, if necessary, in subsequent
periods if actual forfeitures differ from those estimates. Until December 31, 2022, we estimated forfeitures at the time of grant
and would revise our estimate, if necessary, in subsequent periods. As of January 1, 2023, we account for forfeitures as they occur.
The
fair value of Restricted Stock Units, or RSUs, is based on the number of shares granted and market price of the stock on the date of
grant and is recorded as compensation expense ratably over the applicable service period, which is generally four years. Unvested restricted
stock units and vested and unvested stock options are forfeited in the event of termination of employment.
Income
Taxes
Income
taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences
attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective
tax bases, as well as for operating losses and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted
tax rates expected to be applied to taxable income in the years in which those temporary differences are expected to be recovered or
settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes
the enactment date. Deferred tax assets and liabilities are recorded net as long-term on the consolidated balance sheets.
Foreign Currency
Assets and liabilities of non-U.S. operations where
the functional currency is other than the U.S. dollar are translated from the functional currency into U.S. dollars at year end exchange
rates, and revenues and expenses are translated at average rates prevailing during the year. Resulting translation adjustments are accumulated
as part of accumulated other comprehensive income. Transaction gains or losses are recognized in income or loss in the period in which
they occur. The cumulative translation adjustment for the year ended December 31, 2023 was less than $1,000 and therefore not separately
reported on the consolidated financial statements.
A
valuation allowance is recorded when it is more likely than not that some or all of the net deferred tax assets will not be realized.
Accordingly, the Company provides a valuation allowance, if necessary, to reduce net deferred tax assets to the amount that is expected
to be realized.
Tax
positions taken or expected to be taken in the course of preparing the Company’s tax returns are required to be evaluated to determine
whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authority. Tax positions not
deemed to meet a “more-likely-than-not” threshold would be recorded as a tax expense in the current year.
When
necessary, the Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
Net
Loss per Share
Basic
net loss per share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of shares outstanding
during the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by adjusting the weighted-average
number of shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock
method. For purposes of the diluted net loss per share calculation, warrants to purchase common stock and stock options are considered
to be common stock equivalents, but have been excluded from the calculation of diluted net loss per share, as their effect would be anti-dilutive
for all periods presented. Therefore, basic and diluted net loss per share applicable to common stockholders were the same for all periods
presented.
Warrant
Liability
The
Company classifies warrants to purchase shares of its common stock as a liability on its consolidated balance sheets when the warrant
is a free-standing financial instrument that may require the Company to transfer cash consideration upon exercise and that cash transfer
event would be out of the Company’s control. Such a “liability warrant” is initially recorded at fair value on date
of grant using the Black-Scholes model and net of issuance costs, and it is subsequently re-measured to fair value at each subsequent
balance sheet date. Changes in the fair value of the warrant are recognized as a component of other income (expense), net in the consolidated
statements of operations. The Company will continue to adjust the liability for changes in fair value until the earlier of the exercise
or expiration of the warrant.
For
warrants that do not meet the criteria of a liability warrant and are classified on the Company’s consolidated balance sheets as
equity instruments, the Company uses the Black-Scholes model to measure the value of the warrants at issuance and then applies the relative
fair-value of the equity transaction between common stock, preferred stock and warrants. Common stock and equity-classified warrants
each are considered permanent equity.
Concentration
of Credit Risk
Financial
investments that potentially subject the Company to credit risk consist of cash. The Company has all cash at accredited financial institutions.
Bank accounts in the United States are insured by the Federal Deposit Insurance Corporation (FDIC) up to $250,000. The Company does not
believe that it is subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
Recent
Accounting Pronouncements
From
time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies we adopt as of the specified effective
date. Unless otherwise discussed below, we do not believe that the adoption of recently issued standards have or may have a material
impact on our consolidated financial statements.
In
June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial
Instruments (ASU 2016-12). The new standard requires that expected credit losses relating to financial assets measured on an amortized
cost basis and available-for-sale debt securities be recorded through an allowance for credit losses. It also limits the amount of credit
losses to be recognized for available-for-sale debt securities to the amount by which carrying value exceeds fair value and also requires
the reversal of previously recognized credit losses if fair value increases. The Company adopted this standard on January 1, 2023, and
the adoption of ASU 2016-13 did not have a material impact on its consolidated financial statements.
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for all significant accounting policies of the reporting entity. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 235
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//235/tableOfContent
| Name: |
us-gaap_SignificantAccountingPoliciesTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Fair Value Measurements
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Fair Value Disclosures [Abstract] |
|
| Fair Value Measurements |
3.
Fair Value Measurements
Fair
value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal
or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date.
The
Company utilizes a valuation hierarchy for disclosure of the inputs to the valuations used to measure fair value. This hierarchy prioritizes
the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or
liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for
the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial
instrument. Level 3 inputs are unobservable inputs based on the Company’s own assumptions used to measure assets and liabilities
at fair value. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input
that is significant to the fair value measurement.
The Company had no assets or liabilities classified as Level 2 or Level
3 as of December 31, 2023 and 2022. In 2023, the Company had a certificate of deposit which matured in October 2023 with the remaining
$1.2 million released from short-term investments into cash and cash equivalents. The carrying value of financial instruments (consisting
of cash, accounts payable, accrued compensation and accrued expenses) is considered to be representative of their respective fair values
due to the short-term nature of those instruments.
Investment
income is included as interest income in the accompanying consolidated statement of operations for the year ended December 31, 2023.
There
were no transfers between Level 1, Level 2 and Level 3 in either of the years ended December 31, 2023 and December 31, 2022.
|
| X |
- References
+ Details
| Name: |
us-gaap_FairValueDisclosuresAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for the fair value of financial instruments (as defined), including financial assets and financial liabilities (collectively, as defined), and the measurements of those instruments as well as disclosures related to the fair value of non-financial assets and liabilities. Such disclosures about the financial instruments, assets, and liabilities would include: (1) the fair value of the required items together with their carrying amounts (as appropriate); (2) for items for which it is not practicable to estimate fair value, disclosure would include: (a) information pertinent to estimating fair value (including, carrying amount, effective interest rate, and maturity, and (b) the reasons why it is not practicable to estimate fair value; (3) significant concentrations of credit risk including: (a) information about the activity, region, or economic characteristics identifying a concentration, (b) the maximum amount of loss the entity is exposed to based on the gross fair value of the related item, (c) policy for requiring collateral or other security and information as to accessing such collateral or security, and (d) the nature and brief description of such collateral or security; (4) quantitative information about market risks and how such risks are managed; (5) for items measured on both a recurring and nonrecurring basis information regarding the inputs used to develop the fair value measurement; and (6) for items presented in the financial statement for which fair value measurement is elected: (a) information necessary to understand the reasons for the election, (b) discussion of the effect of fair value changes on earnings, (c) a description of [similar groups] items for which the election is made and the relation thereof to the balance sheet, the aggregate carrying value of items included in the balance sheet that are not eligible for the election; (7) all other required (as defined) and desired information. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 820
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482106/820-10-50-2
| Name: |
us-gaap_FairValueDisclosuresTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Prepaid Expenses and Other Current Assets
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Prepaid Expenses And Other Current Assets |
|
| Prepaid Expenses and Other Current Assets |
4.
Prepaid Expenses and Other Current Assets
Prepaid
expenses and other current assets consist of the following:
Schedule
of Prepaid expenses and Other Current Assets
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Deposits | |
$ | — | | |
$ | 20 | |
| Insurance | |
| 8 | | |
| 8 | |
| Prepaid contracts | |
| 79 | | |
| 51 | |
| Total prepaid expenses and other current assets | |
$ | 87 | | |
$ | 79 | |
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosurePrepaidExpensesAndOtherCurrentAssetsAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPrepaid Expenses And Other Current Assets [Text Block]
| Name: |
HRGN_PrepaidExpensesAndOtherCurrentAssetsTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Property, Plant and Equipment, Net
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Property, Plant and Equipment [Abstract] |
|
| Property, Plant and Equipment, Net |
5.
Property, Plant and Equipment, Net
Property,
plant and equipment, net consist of the following:
Schedule
of Property Plant and Equipment Net
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Leasehold improvements | |
$ | 35 | | |
$ | 35 | |
| Furniture, machinery and equipment | |
| 1,414 | | |
| 1,405 | |
| Computer equipment and software | |
| 38 | | |
| 36 | |
| Total property, plant and equipment | |
| 1,487 | | |
| 1,476 | |
| Less: accumulated depreciation | |
| (1,462 | ) | |
| (1,427 | ) |
| Property, plant and equipment, net | |
$ | 25 | | |
$ | 49 | |
Depreciation
expense amounted to approximately $35,000 and $52,000 for the years ended December 31, 2023 and 2022, respectively.
|
| X |
- References
+ Details
| Name: |
us-gaap_PropertyPlantAndEquipmentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for long-lived, physical asset used in normal conduct of business and not intended for resale. Includes, but is not limited to, work of art, historical treasure, and similar asset classified as collections. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//360/tableOfContent
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-6
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-7
| Name: |
us-gaap_PropertyPlantAndEquipmentDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Long-term prepaid contracts
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Long-term Prepaid Contracts |
|
| Long-term prepaid contracts |
6. Long-term prepaid contracts
We have contracted with partners
relating to our clinical trial activities. Upon execution of the contracts, we made initial payments of $1.2
million as deposits recorded as long-term assets and will be applied against final invoices which are more than a year away. The deposits
will be recorded as expense when the clinical trial is substantially completed. Costs for the clinical trial activities throughout
our clinical trial under these contracts are recognized as expense and payable based on costs incurred.
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLongtermPrepaidContractsAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLong Term Prepaid Contracts [Text Block]
| Name: |
HRGN_LongTermPrepaidContractsTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Accrued and Other Current Liabilities
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Payables and Accruals [Abstract] |
|
| Accrued and Other Current Liabilities |
7.
Accrued and Other Current Liabilities
Accrued
and other current liabilities consist of the following:
Schedule
of Accrued and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Advisory costs | |
$ | 325 | | |
$ | 300 | |
| Legal costs | |
| — | | |
| 135 | |
| Audit services | |
| 70 | | |
| 80 | |
| Payroll | |
| 79 | | |
| 55 | |
| Other liabilities | |
| 1 | | |
| 12 | |
| Total expenses | |
$ | 475 | | |
$ | 582 | |
|
| X |
- DefinitionThe entire disclosure for accounts payable, accrued expenses, and other liabilities that are classified as current at the end of the reporting period.
| Name: |
us-gaap_AccountsPayableAccruedLiabilitiesAndOtherLiabilitiesDisclosureCurrentTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Warrant Liability
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Warrant Liability |
|
| Warrant Liability |
8.
Warrant Liability
During
2016 and 2017, the Company closed a sale of shares of the Company’s common stock, the issuance of warrants to purchase shares of
common stock, and the issuance of warrants to the placement agent for each transaction. Due to a cash put provision within the warrant
agreement, which could be enacted in certain change in control events, a liability associated with those 1,044,396 warrants were initially
recorded at fair value and subsequently re-measured each reporting period. The changes in the fair value between issuance and the end
of each reporting period is recorded as a component of other income (expense), net in the consolidated statements of operations.
During
2017, the holders of 952,184 warrants agreed to a modification of the term which removed the cash put provision. The remaining 92,212
warrants continued to be re-measured at each reporting period as long as they were outstanding and un-modified. In February 2022, the
remaining 92,212 warrants expired unexercised.
The
following table presents a reconciliation of the Company’s warrant liabilities for the year ended December 31, 2022:
Schedule
of Warrant Liability
| |
|
Warrant
Liability |
|
| |
|
(in
thousands) |
|
| Balance
as of January 1, 2022 |
|
$ |
2 |
|
| Change
in fair value upon re-measurement |
|
|
(2 |
) |
| Balance
as of December 31, 2022 |
|
$ |
— |
|
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureWarrantLiabilityAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWarrant Liability [Text Block]
| Name: |
HRGN_WarrantLiabilityTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Commitments and Contingencies
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Commitments and Contingencies Disclosure [Abstract] |
|
| Commitments and Contingencies |
9.
Commitments and Contingencies
On
April 14, 2017, representatives for the estate of an individual plaintiff filed a wrongful death complaint with the Suffolk Superior
Court, in the County of Suffolk, Massachusetts, against the Company and other defendants, including Harvard Bioscience, Inc., or HBIO,
the former parent of the Company that spun off the Company in 2013, as well as another third party. The complaint sought payment for
an unspecified amount of damages and alleged that the plaintiff sustained terminal injuries allegedly caused by products provided by
certain of the named defendants and utilized in connection with surgeries performed by third parties in Europe in 2012 and 2013. This
lawsuit related to the Company’s first-generation trachea scaffold technology for which the Company discontinued development in
2014, and not to the Company’s current HRGN Esophageal Implant.
On
April 27, 2022, the Company and HBIO executed a settlement with the plaintiffs (the “Settlement”), which resolves all claims
relating to the litigation. The Settlement resulted in the dismissal with prejudice of the wrongful death claim, and neither the Company
nor HBIO admit any fault or liability in connection with the claim. The Settlement also resolved any and all claims by and between the
parties and the Company’s product liability insurance carriers, which resulted in the dismissal with prejudice of all claims asserted
by or against those carriers, the Company and HBIO.
In
relation to the litigation, the Company paid approximately $5.9 million of aggregate costs related to the lawsuit. As of December 31,
2022, all such lawsuit related costs had been paid or otherwise satisfied as provided below. This aggregate amount included the cost
of legal and related costs incurred by the Company, which consisted of attorneys’ fees and advisor and specialist costs as part
of its defense in this matter. On March 3, 2022, the Company received a cash payment of approximately $0.1 million from Medmarc, the
Company’s insurance carrier. This amount represented a reimbursement of previously incurred legal costs and was recorded as a reduction
to general and administrative expenses during the year ended December 31, 2022.
With
respect to such $5.9 million of costs described above, the Company was required to either pay such costs directly or indemnify HBIO as
to such amounts it incurs. Of such amounts, the Company anticipated that HBIO would pay an aggregate amount of $4.0 million by the end
of the second quarter of 2022. With respect to the indemnification obligation of the Company to HBIO pertaining to such costs, the Company
and HBIO entered into a Preferred Issuance Agreement dated as of April 27, 2022 (the PIA). In connection with the PIA, the Company and
HBIO agreed that once HBIO had paid at least $4.0 million in such costs, to satisfy the Company’s indemnification obligations with
respect thereto, in lieu of paying cash, the Company would issue senior 8% convertible preferred stock to HBIO that will contain terms
as described in the PIA, including the term sheet attached thereto. On June 10, 2022, following the execution of a subscription agreement
and HBIO providing evidence of payment of the requisite $4.0 million amount, the Company issued HBIO 4,000 shares of Series E 8% Convertible
Preferred Stock at a price of $1,000 per share to satisfy the Company’s related indemnification obligations aggregating $4.0 million,
which included the accrual for contingency of $3.3 million and approximately $0.8 million of legal and related costs paid on behalf of
the Company by HBIO previously included in accrued expenses.
From
time to time, the Company may be involved in various claims and legal proceedings arising in the ordinary course of business. Other than
the above matter, there are no such matters pending that the Company expects to be material in relation to its business, financial condition,
results of operations, or cash flows.
We currently have a co-development initiative
with Yale University and the McGowan Institute for Regenerative Medicine at the University of Pittsburgh. We are required to make advance
payments of approximately $130,000 and $61,000, respectively at inception of the contracts. We plan to make these advance payments in
the second quarter of 2024. The universities started preparatory work in 2023 with substantial work to be done in 2024. Either party
can terminate the contract with reasonable notice and any incurred costs will be reimbursed by us to the universities.
|
| X |
- References
+ Details
| Name: |
us-gaap_CommitmentsAndContingenciesDisclosureAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for commitments and contingencies. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 440
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482648/440-10-50-4
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//450/tableOfContent
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 954
-SubTopic 440
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480327/954-440-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 440
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482648/440-10-50-4
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 440
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//440/tableOfContent
| Name: |
us-gaap_CommitmentsAndContingenciesDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Leases
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Leases |
|
| Leases |
10.
Leases
The
Company leases laboratory and office space and certain equipment with remaining terms ranging from 1 year to 3 years.
The
laboratory and office space arrangement is under a sublease that was renewed in December of 2022 and currently extends through May 31,
2024. This lease automatically renews annually for one-year periods unless the Company or the counterparty provides a notice of termination
within one hundred and eighty days prior to May 31st of each year.
All
of the Company’s leases qualify as operating leases. The following table summarizes the presentation of the Company’s operating
leases in its consolidated balance sheets:
Schedule of Operating Leases in Consolidated Balance Sheets
| | |
| |
December 31, | |
| | |
Balance Sheet Classification | |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Assets: | |
| |
| | |
| |
| Operating lease assets | |
Right-of-use asset, net | |
$ | 48 | | |
$ | 147 | |
| Liabilities: | |
| |
| | | |
| | |
| Current portion of operating lease liabilities | |
Current portion of operating lease liabilities | |
| 48 | | |
| 99 | |
| Operating lease liabilities, net of current portion | |
Operating lease liabilities, net of current portion | |
| — | | |
| 48 | |
| Total operating lease liabilities | |
| |
$ | 48 | | |
$ | 147 | |
Cash
paid for leases during each
of the years ended December 31, 2023 and 2022 amounted to approximately $127,000 and $121,000, respectively.
The
weighted average remaining lease terms and weighted average discount rates as of December 31, 2023 and 2022 were as follows:
Schedule
of Weighted Average Lease Term and Discount Rates
| | |
Year ended December 31, | |
| | |
2023 | | |
2022 | |
| Remaining lease term (in years) | |
| 0.48 | | |
| 1.43 | |
| Discount rate | |
| 14.66 | % | |
| 14.74 | % |
The
following table summarizes the effect of lease costs in the Company’s consolidated statements of operations:
Schedule
of Operating Lease Expense Categories in Consolidated Statements of Operations
| | |
| |
For the Year Ended December 31, | |
| | |
| |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Operating lease expense | |
Research and development | |
$ | 68 | | |
$ | 77 | |
| | |
Sales and marketing | |
| 15 | | |
| — | |
| | |
General and administrative | |
| 44 | | |
| 44 | |
| | |
Total | |
$ | 127 | | |
$ | 121 | |
The
minimum lease payments for the next year is as follows:
Schedule
of Minimum Lease Payments
| | |
As of | |
| | |
December 31, 2023 | |
| | |
(in thousands) | |
| 2024 | |
$ | 50 | |
| Total lease payments | |
| 50 | |
| Less: imputed interest | |
| (2 | ) |
| Present value of operating lease liabilities | |
$ | 48 | |
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLeasesAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for operating leases of lessee. Includes, but is not limited to, description of operating lease and maturity analysis of operating lease liability. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//842-20/tableOfContent
| Name: |
us-gaap_LesseeOperatingLeasesTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Income Taxes
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Income Tax Disclosure [Abstract] |
|
| Income Taxes |
11.
Income Taxes
A
reconciliation of taxes utilizing the expected federal tax rate of 21% and the effective tax rate is as follows:
Schedule of Effective Income Tax
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| Computed “expected” income tax benefit | |
| 21.0 | % | |
| 21.0 | % |
| State income tax benefit, net of federal income tax benefit | |
| 6.3 | % | |
| 6.3 | % |
| Tax credits | |
| 0.6 | % | |
| 0.8 | % |
| Change in valuation allowance | |
| (27.9 | )% | |
| (28.1 | )% |
| Total income taxes | |
| — | % | |
| — | % |
The
components of the Company’s deferred tax assets and liabilities are as follows:
Schedule
of Deferred tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Deferred tax assets: | |
| | | |
| | |
| Operating loss and credit carryforwards | |
$ | 19,167 | | |
$ | 20,487 | |
| Capitalized research and development | |
| 1,288 | | |
| 1,083 | |
| Stock-based compensation | |
| 2,504 | | |
| 1,566 | |
| Lease liabilities | |
| 13 | | |
| 40 | |
| Total deferred tax assets | |
| 22,972 | | |
| 23,176 | |
| Less: valuation allowance | |
| (22,959 | ) | |
| (23,136 | ) |
| Deferred tax assets | |
| 13 | | |
| 40 | |
| | |
| | | |
| | |
| Deferred tax liability: | |
| | | |
| | |
| Operating lease assets | |
| (13 | ) | |
| (40 | ) |
| Total deferred tax liability | |
| (13 | ) | |
| (40 | ) |
| Deferred
Tax Net | |
$ | — | | |
$ | — | |
The
Company has recorded a valuation allowance against its deferred tax assets for the years ended December 31, 2023 and 2022, because the
Company’s management believes that it is more likely than not that these assets will not be realized. The valuation allowance decreased
by approximately $0.2
million for the year ended December 31, 2023 and increased by approximately $2.9 million
for the year ended December 31, 2022, respectively, primarily as a result of operating losses generated with no corresponding financial
statement benefit.
As
of December 31, 2023, the Company had federal net operating loss carryforwards, or NOLs, of approximately $68.7 million to offset future
federal taxable income and state NOLs of approximately $68.1 million to offset future state taxable income. The federal and state NOLs
generated for annual periods prior to January 1, 2018 begin to expire in 2033. The Company’s federal NOL generated for the years
ended December 31, 2018 through December 31, 2023, which amount to $42.3 million, can be carried forward indefinitely, however, are limited
to be utilized to offset 80% of taxable income in each successive year. As of December 31, 2023, the Company also has federal and state
tax research and development credit carryforwards of approximately $1.5 million and $1.0 million, respectively, to offset future income
taxes. The federal and state research and development tax credit carryforwards begin to expire in 2033 and 2029, respectively.
Under
the provisions of the Internal Revenue Code, the net operating loss and tax credit carryforwards are subject to review and possible adjustment
by the Internal Revenue Service and state tax authorities. Net operating loss and tax credit carryforwards may become subject to an annual
limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period
in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code, respectively, as well as similar state provisions.
This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount
of the annual limitation is determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership
changes may further affect the limitation in future years. The Company has recently completed several equity financings transactions
which have either individually or cumulatively resulted in a change in control as defined by Sections 382 and 383 of the Internal Revenue
Code or could result in a change in control in the future. The Company does not believe the impact of any limitation on the use of its
net operating loss or credit carryforwards will have a material impact on the Company’s consolidated financial statements since
the Company has a full valuation allowance against its net deferred tax assets due to the uncertainty regarding future taxable income
for the foreseeable future.
For
all years through December 31, 2023, the Company generated research credits but has not conducted a study to document the qualified activities.
This study may result in an adjustment to the Company’s research and development credit carryforwards; however, until a study is
completed, and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been
provided against the Company’s research and development credits and, if an adjustment is required, this adjustment would be offset
by an adjustment to the deferred tax asset established for the research and development credit carryforwards and the valuation allowance.
Harvard
Bioscience received a Supplemental Ruling to the Private Letter Ruling dated March 22, 2013 from the IRS to the effect that, among other
things, the Separation and Distribution by Harvard Bioscience will qualify as a transaction that is tax-free for U.S. federal income
tax purposes under Section 355 and 368(a)(1)(D) of the Internal Revenue Code continuing in effect. The private letter and supplemental
rulings and the tax opinion that Harvard Bioscience received from legal counsel to Harvard Bioscience rely on certain representations,
assumptions and undertakings, including those relating to the past and future conduct of the Harvard Apparatus Regenerative Technology
business, and neither the private letter and supplemental rulings nor the opinion would be valid if such representations, assumptions
and undertakings were incorrect. Moreover, the private letter and supplemental rulings do not address all the issues that are relevant
to determining whether the Distribution will qualify for tax-free treatment. Notwithstanding the private letter and supplemental rulings
and opinion, the IRS could determine the Distribution should be treated as a taxable transaction for U.S. federal income tax purposes
if, among other reasons, it determines any of the representations, assumptions or undertakings that were included in the request for
the private letter and supplemental rulings are false or have been violated or if it disagrees with the conclusions in the opinion that
are not covered by the IRS ruling.
To
preserve the tax-free treatment to Harvard Bioscience of the Separation and Distribution, for the two-year period following the Distribution,
which such period ended November 1, 2015, the Company was limited, except in specified circumstances, from entering into certain transactions
pursuant to which all or a portion of the Company’s stock would be acquired, whether by merger or otherwise; issuing equity securities
beyond certain thresholds; repurchasing the Company’s common stock; and ceasing to actively conduct the Company’s regenerative
medicine business. In addition, at all times, including during and following such two-year period, the Company may not take or fail to
take any other action that prevents the Separation and Distribution and related transactions from being tax-free.
If
the Distribution fails to qualify for tax-free treatment, in general, Harvard Bioscience would be subject to tax as if it had sold the
Company’s common stock in a taxable sale for its fair market value, and Harvard Bioscience stockholders who received shares of
Harvard Apparatus Regenerative Technology common stock in the Distribution would be subject to tax as if they had received a taxable
Distribution equal to the fair market value of such shares.
Under
the tax sharing agreement between Harvard Bioscience and the Company, the Company would generally be required to indemnify Harvard Bioscience
against any tax resulting from the Distribution to the extent that such tax resulted from (i) an acquisition of all or a portion of the
Company’s stock or assets, whether by merger or otherwise, (ii) other actions or failures to act by the Company, or (iii) any of
the Company’s representations or undertakings being incorrect or violated. The Company’s indemnification obligations to Harvard
Bioscience and its subsidiaries, officers and directors are not limited by any maximum amount. If the Company is required to indemnify
Harvard Bioscience or such other persons under the circumstances set forth in the tax sharing agreement, the Company may be subject to
substantial liabilities.
All
deferred tax assets prior to the Separation remained with Harvard Bioscience.
The
Company has determined that any uncertain tax positions would have no material impact on the consolidated financial statements of the
Company and there are no unrecognized tax benefits or related interest and penalties accrued for the period for the years ended December
31, 2023 and 2022.
The
Company is subject to U.S. federal income tax and Massachusetts state income tax. The statute of limitations for assessment by the IRS
and state tax authorities is open for all periods from inception through December 31, 2022; currently, no federal or state income tax
returns are under examination by the respective taxing authorities.
On
March 27, 2020, the Coronavirus Aid, Relief, and Economic Security, or CARES, Act was signed into law making several changes to the Internal
Revenue Code. The changes include but are not limited to increasing the limitation on the amount of deductible interest expense, allowing
companies to carryback certain net operating losses, and increasing the amount of net operating loss carryforwards that corporations
can use to offset taxable income. The tax law changes in the CARES Act did not have a material impact on the Company’s income tax
provision.
|
| X |
- DefinitionThe entire disclosure for income taxes. Disclosures may include net deferred tax liability or asset recognized in an enterprise's statement of financial position, net change during the year in the total valuation allowance, approximate tax effect of each type of temporary difference and carryforward that gives rise to a significant portion of deferred tax liabilities and deferred tax assets, utilization of a tax carryback, and tax uncertainties information. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480990/946-20-50-13
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(h)(2))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//740/tableOfContent
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-14
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 21
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-21
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 270
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482526/740-270-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-17
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB TOPIC 6.I.5.Q1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479360/740-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 11.C)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479360/740-10-S99-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482603/740-30-50-2
| Name: |
us-gaap_IncomeTaxDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Employee Benefit Plan
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Retirement Benefits [Abstract] |
|
| Employee Benefit Plan |
12.
Employee Benefit Plan
The
Company sponsors a retirement plan for its U.S. employees, which includes an employee savings plan established under Section 401(k) of
the U.S. Internal Revenue Code, or the 401(k) Plan. The 401(k) Plan covers substantially all full-time employees who meet certain eligibility
requirements. Contributions to the retirement plan are at the discretion of management. The Company’s matching contributions to
the plan were approximately $66,000 and $35,000 for the years ended December 31, 2023 and 2022, respectively.
|
| X |
- DefinitionThe entire disclosure for an entity's employee compensation and benefit plans, including, but not limited to, postemployment and postretirement benefit plans, defined benefit pension plans, defined contribution plans, non-qualified and supplemental benefit plans, deferred compensation, share-based compensation, life insurance, severance, health care, unemployment and other benefit plans. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 710
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//710/tableOfContent
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 712
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//712/tableOfContent
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 715
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//715/tableOfContent
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 718
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//718/tableOfContent
| Name: |
us-gaap_CompensationAndEmployeeBenefitPlansTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_CompensationAndRetirementDisclosureAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Series E Convertible Preferred Stock
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Series E Convertible Preferred Stock |
|
| Series E Convertible Preferred Stock |
13.
Series E Convertible Preferred Stock
On
April 28, 2022, the Company entered into a Preferred Issuance Agreement, or PIA, with Harvard Bioscience, Inc., or HBIO, dated as of
April 27, 2022. Pursuant to the PIA, the Company and HBIO agreed that once HBIO has paid at least $4.0 million in certain settlement
and related legal expenses, to satisfy the Company’s indemnification obligations with respect thereto, in lieu of paying cash,
the Company would issue senior convertible preferred stock to HBIO that will contain terms as described in the PIA.
On
June 10, 2022, following the execution of a subscription agreement and HBIO providing evidence of payment of the requisite $4.0 million
amount, the Company issued HBIO 4,000 shares of Series E Convertible Preferred Stock, or Series E Preferred, at a price of $1,000 per
share to satisfy the Company’s related indemnification obligations pertaining to the $4.0 million, in lieu of paying cash.
On
January 18, 2023, HBIO converted 200 Series E Preferred Shares with accrued dividends of $9,545 into 31,933 shares of common stock.
In
connection with the private placement, as of April 12, 2023, the Company had received $6.0 million in aggregate proceeds in such private
placement. The private placement resulted in gross proceeds of at least $4.0 million which triggered the mandatory conversion of all
the Company’s outstanding Series E Preferred Stock and related accrued dividends into shares of common stock at a conversion price
of $6.00 per share. The conversion resulted in 674,693 shares of common stock being issued to the holder of the Series E Preferred Stock.
Following such conversion, there are no shares of Series E Preferred Stock outstanding.
There
were no
shares of any of the classes of preferred stock outstanding as of December 31, 2023. There were no changes to authorized shares for
the years ending December 31, 2022 and 2023. Authorized shares for each preferred stock class are as follows:
Schedule
of Categories of Preferred Stock
| | |
Authorized | |
| Undesignated Preferred Stock | |
| 979,000 | |
| Series B Convertible Preferred Stock | |
| 1,000,000 | |
| Series C Convertible Preferred Stock | |
| 4,000 | |
| Series D Convertible Preferred Stock | |
| 12,000 | |
| Series E Convertible Preferred Stock | |
| 5,000 | |
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureSeriesEConvertiblePreferredStockAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTemporary Equity Disclosure [Text Block]
| Name: |
HRGN_TemporaryEquityDisclosureTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Common Stock
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Equity [Abstract] |
|
| Common Stock |
14.
Common Stock
The
Company has 60,000,000 shares authorized as of December 31, 2023 and 40,961,765 shares of common stock available for issuance.
The
following represent the Company’s common stock transactions during December 31, 2023 and 2022:
2023
Capital Transactions
On
April 12, 2023 and on March 31, 2023, the Company entered into Securities Purchase Agreements, each a Purchase Agreement, with new and
existing investors, the Investors, pursuant to which the Investors agreed to purchase in a private placement an aggregate of 1,000,967
shares of common stock for the aggregate purchase price of approximately $6 million with a purchase price per unit of $6.00.
2022
Capital Transactions
On
May 12, 2022, the Company entered into Securities Purchase Agreements, each a Purchase Agreement, with new and existing investors, the
Investors, pursuant to which the Investors agreed to purchase in a private placement an aggregate of 854,771 shares of common stock and
warrants to purchase 427,390 shares of common stock, subject to adjustment as provided in the warrant agreement, the Warrants, for the
aggregate purchase price of approximately $5.1 million with a purchase price per unit of $5.92, the Private Placement. Each unit consisted
of one share of common stock and a warrant to purchase one half of one share of common stock, subject to adjustment, as provided in the
Warrants. The Company received an aggregate of $5.1 million gross and net proceeds from the Private Placement by May 16, 2022.
The
$5.1 million of gross and net proceeds were allocated $3.6 million and $1.5 million to the common stock and warrants, respectively.
The Company classified these warrants on its consolidated balance sheets as equity as the warrants do not have any redemption features
nor a right to put for cash that is outside the control of the Company, and valued using the Black-Scholes model based on the following
weighted average assumptions:
Schedule
of Black-Scholes Model Based on Weighted Average Assumptions
| Risk-free interest rate | |
| 2.81 | % |
| Expected volatility | |
| 127.36 | % |
| Expected term | |
| 5 years | |
| Expected dividend yield | |
| — | |
| Exercise price | |
$ | 8.88 | |
| Market value of common stock | |
$ | 5.50 | |
In
June 2022, the Company issued 4,000 shares of Series E Convertible Preferred Stock at a price of $1,000 per share to satisfy certain
indemnification obligations in the amount of $4.0 million, in lieu of paying cash. The Company issued an aggregate of 180 shares of Series
E Convertible Preferred Stock relating to accrued dividends during the year ended December 31, 2022.
Warrant
to purchase common stock activity for the year ended December 31, 2022 was as follows:
Schedule of Warrant to Purchase Common Stock
| | |
| | |
Weighted-average | |
| | |
Amount | | |
exercise price | |
| Outstanding at January 1, 2022 | |
| 2,501,419 | | |
$ | 4.35 | |
| Issued | |
| 427,390 | | |
| 8.88 | |
| Exercised | |
| (775,000 | ) | |
| 7.17 | |
| Expired | |
| (1,040,187 | ) | |
| 7.59 | |
| Outstanding at December 31, 2022 | |
| 1,113,622 | | |
| 4.69 | |
| Outstanding at December 31, 2023 | |
| 1,113,622 | | |
| 4.69 | |
There was no warrant activity during the year ended December 31, 2023.
Employee
Stock Purchase Plan
The
Company maintains the 2013 Employee Stock Purchase Plan, or the ESPP Plan, whereas participating employees can authorize the Company
to withhold a portion of their base pay during consecutive six-month payment periods for the purchase of shares of the Company’s
common stock. At the conclusion of the period, participating employees can purchase shares of the Company’s common stock at 85%
of the lower of the fair market value of the Company’s common stock at the beginning or end of the period. Shares are issued under
the plan for the six-month periods ending June 30 and December 31. Under this plan, 7,500 shares of common stock are authorized for issuance
of which 4,534 shares have been issued as of December 31, 2023. There are 2,966 shares available for issuance as of December 31, 2023
and December 31, 2022. There was no ESPP Plan activity in 2023 or 2022.
|
| X |
- References
+ Details
| Name: |
us-gaap_EquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for equity. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (h)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 235
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481062/946-235-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 235
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481062/946-235-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-6
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480237/815-40-50-6
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(e)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 10: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//505/tableOfContent
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (g)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 16
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-16
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
| Name: |
us-gaap_StockholdersEquityNoteDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Share-based Compensation
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Share-Based Payment Arrangement [Abstract] |
|
| Share-based Compensation |
15.
Share-based Compensation
Harvard
Apparatus Regenerative Technology Amended and Restated Equity Incentive Plan
The
Company maintains the Amended and Restated Equity Incentive Plan, or the Plan, for the benefit of certain officers, employees, non-employee
directors, and other key persons (including consultants and advisory board members). All options and awards granted under the Plan consist
of the Company’s shares of common stock. The Company’s policy is to issue stock available from its registered but unissued
stock pool through its transfer agent to satisfy stock option exercises and the vesting of restricted stock units. The vesting period
for awards is generally four years and the contractual life is ten years. Canceled and forfeited options and awards are available to
be reissued under the Plan.
As
of December 31, 2023, the Company’s Plan has 9,098,000
authorized shares to be issued under the Plan. There are 5,034,760
shares available for issuance under the Plan as of December 31, 2023.
Stock
option activity under the Plan for the years ended December 31, 2022 and 2023 was as follows:
Schedule of Stock Option Activity
| | |
Amount | | |
Weighted-average exercise price | | |
Weighted-average contractual life (years) | | |
Aggregate intrinsic value (in thousands) | |
| Outstanding at January 1, 2022 | |
| 2,332,603 | | |
$ | 3.93 | | |
| 8.30 | | |
$ | 294 | |
| Granted | |
| 334,418 | | |
| 4.84 | | |
| | | |
| | |
| Canceled / forfeited | |
| (150,097 | ) | |
| 3.39 | | |
| | | |
| | |
| Outstanding at December 31, 2022 | |
| 2,516,924 | | |
| 3.95 | | |
| 7.68 | | |
| 6,917 | |
| Granted | |
| 2,130,007 | | |
| 5.81 | | |
| | | |
| | |
| Exercised | |
| (65,264 | ) | |
| 2.29 | | |
| | | |
| | |
| Canceled / forfeited | |
| (604,378 | ) | |
| 6.13 | | |
| | | |
| | |
| Outstanding at December 31, 2023 | |
| 3,977,289 | | |
$ | 4.64 | | |
| 7.7 | | |
$ | 5,728 | |
| Options exercisable at December 31, 2023 | |
| 2,281,655 | | |
$ | 4.64 | | |
| 7.24 | | |
$ | 4,209 | |
| Options vested or expected to vest at December 31, 2023 | |
| 3,877,871 | | |
$ | 4.69 | | |
| 7.7 | | |
$ | 5,522 | |
The
Company’s outstanding stock options include 773,195 performance-based awards that have vesting provisions subject to the achievement
of certain business milestones. Total unrecognized compensation expense for the remaining performance-based awards is approximately $2.8
million. No expense has been recognized for these awards as of December 31, 2023 given that the milestone achievements for these awards
have not yet been deemed probable for accounting purposes.
Aggregate
intrinsic value for outstanding options for the year ended December 31, 2023 was approximately $5.7
million and calculated as the difference between the Company’s closing stock price of $4.99
per share as of December 29, 2023 and the weighted average exercise price of $4.64.
As of December 31, 2023, unrecognized compensation cost related to unvested non-performance-based awards amounted to $3.9
million, which will be recognized over a weighted-average period of 2.2
years.
The
weighted average assumptions for valuing the Company’s stock options granted were as follows:
Schedule of Weighted Average Assumptions
| | |
Year Ended December 31, | |
| | |
2023 | | |
2022 | |
| Risk-free interest rate | |
| 3.82 | % | |
| 2.74 | % |
| Expected volatility | |
| 125.35 | % | |
| 123.61 | % |
| Expected term (in years) | |
| 5.8 years | | |
| 5.8 years | |
| Expected dividend yield | |
| — | % | |
| — | % |
The
grant date fair value of stock options is estimated using the Black-Scholes option pricing model that takes into account the fair value
of its common stock, the exercise price, the expected life of the option, the expected volatility of its common stock, expected dividends
on its common stock, and the risk-free interest rate over the expected life of the option. The risk-free interest rate assumption is
based upon observed treasury bill interest rates (risk-free) appropriate for the expected term of the Company’s employee stock
options. The computation of expected volatility is based on the historical volatility of the Company’s common stock. The simplified
method of estimating expected term was used. The Company has not paid and do not anticipate paying cash dividends on the Company’s
shares of common stock; therefore, the expected dividend yield is assumed to be zero.
The
weighted average estimated fair value of stock options granted using the Black-Scholes model was $5.12 and $4.23 per share for the years
ended December 31, 2023 and 2022, respectively.
The
Company also estimated the fair value of non-employee share options using the Black-Scholes option pricing model reflecting the same
assumptions as applied to employee and director options in each of the reporting periods, other than the expected life, which is assumed
to be the remaining contractual life of the options.
Share-based
compensation expense related to the Plan for the years ended December 31, 2023 and 2022 was allocated as follows:
Schedule
of Share-based Compensation Expense
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Research and development | |
$ | 327 | | |
$ | 284 | |
| Selling, general and administrative | |
| 3,134 | | |
| 747 | |
| Total stock-based compensation | |
$ | 3,461 | | |
$ | 1,031 | |
|
| X |
- DefinitionThe entire disclosure for share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//718/tableOfContent
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (l)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_DisclosureOfCompensationRelatedCostsShareBasedPaymentsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Net Loss per Share
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Earnings Per Share [Abstract] |
|
| Net Loss per Share |
16.
Net Loss per Share
Basic
and diluted net loss per share was calculated as follows:
Schedule of Basic and Diluted Net Loss Per Share
| | |
2023 | | |
2022 | |
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands, except shares and per share data) | |
| Net loss | |
$ | (8,945 | ) | |
$ | (6,073 | ) |
| Preferred stock dividends | |
| (77 | ) | |
| (180 | ) |
| Net loss attributable to common stockholders | |
| (9,022 | ) | |
| (6,253 | ) |
| Basic and diluted weighted average common shares outstanding | |
| 13,455,666 | | |
| 11,349,610 | |
| Basic and diluted net loss per share attributable to common stockholders | |
$ | (0.67 | ) | |
$ | (0.55 | ) |
The
Company’s potentially dilutive securities, which include stock options, unvested restricted common stock units and warrants, have
been excluded from the computation of diluted net loss per share whenever the effect of including them would be to reduce the net loss
per share. In periods where there is a net loss, the weighted average number of common shares outstanding used to calculate both basic
and diluted net loss per share attributable to common stockholders is the same.
The
following potential common shares were excluded from the calculation of diluted net loss per share attributable to common stockholders
for the years ended December 31, 2023 and 2022 because including them would have had an anti-dilutive effect:
Schedule
of Antidilutive Securities Excluded from Computation of Earnings per Share
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| Warrants to purchase common stock | |
| 1,113,622 | | |
| 1,113,622 | |
| Options to purchase common stock | |
| 3,977,289 | | |
| 2,516,924 | |
| Series E convertible preferred stock | |
| — | | |
| 619,259 | |
| Total | |
| 5,090,911 | | |
| 4,249,805 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_EarningsPerShareAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for earnings per share. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//260/tableOfContent
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-3
| Name: |
us-gaap_EarningsPerShareTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Segments and Geographical Information
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Segment Reporting [Abstract] |
|
| Segments and Geographical Information |
17.
Segments and Geographical Information
The
Company’s chief operating decision maker is its Chief Executive Officer. The Company’s chief operating decision maker evaluates
the operating results of the Company’s reportable segments based on revenues and net income (loss).
The Company has two operating and reportable segments: i) Regenerative Biotech
focused on the development of regenerative medicine treatments with operations currently in the United States and ii) Longevity Products
relating to longevity products with operations currently in Asia. The following table presents the Company’s reportable
segment results for the year ended 2023:
Schedule of Reportable Segments
| | |
| | | |
| | |
|
| | |
| | |
Regenerative Biotech | | |
Longevity Products | |
|
Total | |
| 2023 | |
| | | |
| | |
|
| | |
| Revenues | |
$ | — | | |
$ | 103 | |
|
$ | 103 | |
| Net loss | |
| (8,677 | ) | |
| (268 | ) |
|
| (8,945 | ) |
| Total assets | |
| 2,426 | | |
| 188 | |
|
| 2,614 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_SegmentReportingAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for reporting segments including data and tables. Reportable segments include those that meet any of the following quantitative thresholds a) it's reported revenue, including sales to external customers and intersegment sales or transfers is 10 percent or more of the combined revenue, internal and external, of all operating segments b) the absolute amount of its reported profit or loss is 10 percent or more of the greater, in absolute amount of 1) the combined reported profit of all operating segments that did not report a loss or 2) the combined reported loss of all operating segments that did report a loss c) its assets are 10 percent or more of the combined assets of all operating segments. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 42
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-42
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 40
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-40
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//280/tableOfContent
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 26
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-26
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 34
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-34
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 21
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-21
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 21
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-21
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (e)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
| Name: |
us-gaap_SegmentReportingDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Subsequent Events
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Subsequent Events [Abstract] |
|
| Subsequent Events |
18.
Subsequent Events
In March 2024, the Company received cash deposits
in escrow of approximately $0.3 million from a group of prospective investors pertaining to a potential private placement transaction.
These funds remain the respective investor’s property and are being held by the Company in its bank account with Bank of America
until the execution of a common stock purchase agreement.
On
February 1, 2024, the Company entered into a loan arrangement with Junli He, the Chairman and Chief Executive Officer of the Company
(the “Lender”), pursuant to which the Lender has agreed to loan the Company an aggregate amount of $500,000 as evidenced
by a Bridge Note executed by the Company in favor of, and accepted by, the Lender (the “Bridge Note”).
The
Bridge Note accrues interest at an annual fixed rate of 8%, and the principal amount thereof will be due and payable in full, together
with all accrued and unpaid interest thereon, on the earlier to occur of a) the closing date (or later date of capital being provided
pertaining to such continued offering that the following threshold is tripped) of the Company’s next capital raise that includes
gross proceeds of at least $5,000,000 or b) February 1, 2025. The Bridge Note provides for optional conversion at the discretion of the
Lender, contains covenants, and provides for certain events of default including if the Company fails to pay when due any amount owed
thereunder, fails to comply with any agreement, covenant, condition, provision or term contained therein and other customary events of
default.
|
| X |
- References
+ Details
| Name: |
us-gaap_SubsequentEventsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for significant events or transactions that occurred after the balance sheet date through the date the financial statements were issued or the date the financial statements were available to be issued. Examples include: the sale of a capital stock issue, purchase of a business, settlement of litigation, catastrophic loss, significant foreign exchange rate changes, loans to insiders or affiliates, and transactions not in the ordinary course of business. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//855/tableOfContent
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483399/855-10-50-2
| Name: |
us-gaap_SubsequentEventsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Summary of Significant Accounting Policies (Policies)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Principles of Consolidation |
Principles
of Consolidation
The
consolidated financial statements include the accounts of Harvard Apparatus Regenerative Technology, Inc. (Regenerative Biotech) and its three
wholly-owned subsidiaries, Harvard Apparatus Regenerative Technology Limited (Hong Kong), Harvard Apparatus Regenerative Technology
(Hangzhou) Limited (China) and Harvard Apparatus Regenerative Technology GmbH (Germany). All intercompany balances and transactions
have been eliminated in consolidation.
|
| Use of Estimates |
Use
of Estimates
The
process of preparing consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Such estimates include, but are not
limited to, share-based compensation, valuation of warrant liability, accrued expenses and the valuation allowance for deferred income
taxes. Actual results could differ from those estimates.
|
| Revenue |
Revenue
We
recognize revenue in accordance with FASB ASC 606, Revenue from Contracts with Customers. We offer consumer products primarily
through a third-party online store. Revenue is recognized at a point in time when control of the goods is transferred to the customer,
which generally occurs upon the delivery to the customer. For any company direct sales to customers, revenue is recognized at a point
in time upon shipment of product or hand-delivery to customer. Revenue also excludes any amounts collected on behalf of third parties,
including sales and indirect taxes.
We
identify a performance obligation as distinct if both the following criteria are true: the customer can benefit from the good or service
either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer
the good or service to the customer is separately identifiable from other promises in the contract. Determining the standalone selling
price (“SSP”) and allocation of consideration from a contract to the individual performance obligations, and the appropriate
timing of revenue recognition, is the result of significant qualitative and quantitative judgments. Management considers a variety of
factors such as historical sales, usage rates, costs, and expected margin, which may vary over time depending upon the unique facts and
circumstances related to each performance obligation in making these estimates. While changes in the allocation of the SSP between performance
obligations will not affect the amount of total revenue recognized for a particular contract, any material changes could impact the timing
of revenue recognition, which would have a material effect on our financial position and result of operations. This is because the contract
consideration is allocated to each performance obligation, delivered or undelivered, at the inception of the contract based on the SSP
of each distinct performance obligation.
|
| Cost of Sales |
Cost
of Sales
Cost
of sales primarily consists of the purchase price of consumer products, taxes, inbound and outbound shipping costs. Shipping costs to
receive products from our suppliers are recognized as cost of sales when incurred. E-commerce processing and related transaction costs,
including those associated with seller transactions, are classified in sales and marketing on our consolidated statements of
operations.
|
| Research and Development |
Research
and Development
Research
and development costs are expensed as incurred.
|
| Sales and Marketing |
Sales
and Marketing
Sales
and marketing costs include advertising and payroll and related expenses for personnel engaged in marketing and selling activities.
|
| General and Administrative |
General
and Administrative
General
and administrative expenses primarily consist of costs for corporate functions, including payroll and related expenses; facilities and
equipment expenses, such as depreciation and amortization expense and rent; and professional fees.
|
| Segment Information |
Segment Information
The
Company manages its operations as two separate operating segments for the purposes of assessing performance and making operating
decisions. The Company has one operating unit focused on the development and commercialization of therapies to cure patients of
cancers, injuries, and birth defects of the gastro-intestinal tract and the airways. The other operating unit is focused on personal
healthcare through longevity dietary supplements. We have determined that our chief executive officer is the chief operating
decision maker (CODM). The CODM reviews financial information presented by operating unit. Resource allocation decisions are
made by the CODM based on operating unit results.
|
| Cash and Cash Equivalents |
Cash
and Cash Equivalents
The
Company considers all highly liquid investments with a maturity of three months or less at the date of purchase to be cash
equivalents. The Company currently invests available cash in money market funds. As of December 31, 2023, the Company had
approximately $111,000
of cash equivalents in a money market fund.
|
| Accounts Receivable |
Accounts Receivable
Allowances
are provided for estimated amounts of accounts receivable which may not be collected. At
December 31, 2023,
we determined that no allowance against accounts receivable was necessary.
|
| Inventory |
Inventory
Inventory,
consisting of products available for sale, are primarily accounted for using the first-in, first-out method, and are valued at the lower
of cost or net realizable value.
We
maintain ownership of our inventory at the third-party warehouse, regardless of whether fulfillment is provided by us or the third-party
e-commerce seller, and therefore these products are included in our inventories.
|
| Deferred Financing Costs |
Deferred Financing Costs
We capitalized costs relating to a
registered offering that we postponed in 2023 but expect to resume in the near future. The costs include payments made to attorneys,
accountants, regulators and consultants. Once we complete the registered offering, the deferred financing costs will be reclassified
to stockholders’ equity (deficit) on the consolidated balance sheets to offset the proceeds from the registered offering.
|
| Long-term prepaid contracts |
Long-term
prepaid contracts
We
have contracted with partners relating to our clinical trial activities. Upon execution of the contracts, we made initial payments
of $1.2
million as deposits recorded as long-term assets and will be applied against final invoices which are more than a year away. The deposits will be recorded as expense when the clinical trial is substantially
completed. Costs for the clinical trial activities throughout our clinical trial under these contracts are recognized as expense and payable
based on costs incurred.
|
| Property, Plant and Equipment |
Property,
Plant and Equipment
Property,
plant and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets
as follows:
Schedule of Property Plant and Equipment Estimated Useful Lives
| Leasehold improvements | |
Shorter of expected useful life
or lease term |
| Computer equipment and software | |
3 years |
| Furniture, machinery and equipment | |
5-7 years |
Maintenance
and repairs are charged to expense as incurred, while any additions or improvements are capitalized.
|
| Impairment of Long-Lived Assets |
Impairment
of Long-Lived Assets
Assessments
of long-lived assets and the remaining useful lives of such long-lived assets are reviewed for impairment whenever a triggering event
occurs or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. An asset, or group of assets,
are considered to be impaired when the undiscounted estimated net cash flows expected to be generated by the asset, or group of assets,
are less than its carrying amount. The impairment recognized is the amount by which the carrying amount exceeds the fair market value
of the impaired asset, or group of assets, based on the present value of the expected future cash flows associated with the use of the
asset. Through December 31, 2023, no such impairment charges have been recorded.
|
| Share-based Compensation |
Share-based
Compensation
The
Company measures all stock options and restricted stock awards granted to employees, directors and non-employees based on the fair value
on the date of the grant and recognizes compensation expense of those awards, net of forfeitures, over the requisite vesting period,
which is generally the service period of the respective award. Generally, the Company issues stock options and restricted stock awards
with only service-based vesting conditions on a straight-line basis over the requisite service period for the entire award (that is,
over the requisite service period of the last separately vesting portion of the award). Expense on share-based awards for which vesting
is performance or milestone based is recognized on a straight-line basis from the date when it is determined that the achievement of
the milestone is probable to the vesting/milestone achievement date.
The
Company elected to use the Black-Scholes option-pricing model for the valuation of stock-based payment awards. The determination of the
fair value of stock-based payment awards is determined on the date of grant using the Black-Scholes option-pricing model which is affected
by the market price as well as assumptions regarding a number of subjective variables. These variables include, but are not limited to,
its expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. When
performance-based grants are issued, the Company recognizes no expense until achievement of the performance requirement is deemed probable.
Share-based
compensation expense is based on awards ultimately expected to vest and has been reduced for annualized estimated forfeiture where the
minimum amount of expense recorded is at least equal to the percent of an award vested. Forfeitures are estimated based on historical
experience and weighting of various employee classes under the respective plan at the time of grant and revised, if necessary, in subsequent
periods if actual forfeitures differ from those estimates. Until December 31, 2022, we estimated forfeitures at the time of grant
and would revise our estimate, if necessary, in subsequent periods. As of January 1, 2023, we account for forfeitures as they occur.
The
fair value of Restricted Stock Units, or RSUs, is based on the number of shares granted and market price of the stock on the date of
grant and is recorded as compensation expense ratably over the applicable service period, which is generally four years. Unvested restricted
stock units and vested and unvested stock options are forfeited in the event of termination of employment.
|
| Income Taxes |
Income
Taxes
Income
taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences
attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective
tax bases, as well as for operating losses and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted
tax rates expected to be applied to taxable income in the years in which those temporary differences are expected to be recovered or
settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes
the enactment date. Deferred tax assets and liabilities are recorded net as long-term on the consolidated balance sheets.
|
| Foreign Currency |
Foreign Currency
Assets and liabilities of non-U.S. operations where
the functional currency is other than the U.S. dollar are translated from the functional currency into U.S. dollars at year end exchange
rates, and revenues and expenses are translated at average rates prevailing during the year. Resulting translation adjustments are accumulated
as part of accumulated other comprehensive income. Transaction gains or losses are recognized in income or loss in the period in which
they occur. The cumulative translation adjustment for the year ended December 31, 2023 was less than $1,000 and therefore not separately
reported on the consolidated financial statements.
A
valuation allowance is recorded when it is more likely than not that some or all of the net deferred tax assets will not be realized.
Accordingly, the Company provides a valuation allowance, if necessary, to reduce net deferred tax assets to the amount that is expected
to be realized.
Tax
positions taken or expected to be taken in the course of preparing the Company’s tax returns are required to be evaluated to determine
whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authority. Tax positions not
deemed to meet a “more-likely-than-not” threshold would be recorded as a tax expense in the current year.
When
necessary, the Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
|
| Net Loss per Share |
Net
Loss per Share
Basic
net loss per share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of shares outstanding
during the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by adjusting the weighted-average
number of shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock
method. For purposes of the diluted net loss per share calculation, warrants to purchase common stock and stock options are considered
to be common stock equivalents, but have been excluded from the calculation of diluted net loss per share, as their effect would be anti-dilutive
for all periods presented. Therefore, basic and diluted net loss per share applicable to common stockholders were the same for all periods
presented.
|
| Warrant Liability |
Warrant
Liability
The
Company classifies warrants to purchase shares of its common stock as a liability on its consolidated balance sheets when the warrant
is a free-standing financial instrument that may require the Company to transfer cash consideration upon exercise and that cash transfer
event would be out of the Company’s control. Such a “liability warrant” is initially recorded at fair value on date
of grant using the Black-Scholes model and net of issuance costs, and it is subsequently re-measured to fair value at each subsequent
balance sheet date. Changes in the fair value of the warrant are recognized as a component of other income (expense), net in the consolidated
statements of operations. The Company will continue to adjust the liability for changes in fair value until the earlier of the exercise
or expiration of the warrant.
For
warrants that do not meet the criteria of a liability warrant and are classified on the Company’s consolidated balance sheets as
equity instruments, the Company uses the Black-Scholes model to measure the value of the warrants at issuance and then applies the relative
fair-value of the equity transaction between common stock, preferred stock and warrants. Common stock and equity-classified warrants
each are considered permanent equity.
|
| Concentration of Credit Risk |
Concentration
of Credit Risk
Financial
investments that potentially subject the Company to credit risk consist of cash. The Company has all cash at accredited financial institutions.
Bank accounts in the United States are insured by the Federal Deposit Insurance Corporation (FDIC) up to $250,000. The Company does not
believe that it is subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
|
| Recent Accounting Pronouncements |
Recent
Accounting Pronouncements
From
time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies we adopt as of the specified effective
date. Unless otherwise discussed below, we do not believe that the adoption of recently issued standards have or may have a material
impact on our consolidated financial statements.
In
June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial
Instruments (ASU 2016-12). The new standard requires that expected credit losses relating to financial assets measured on an amortized
cost basis and available-for-sale debt securities be recorded through an allowance for credit losses. It also limits the amount of credit
losses to be recognized for available-for-sale debt securities to the amount by which carrying value exceeds fair value and also requires
the reversal of previously recognized credit losses if fair value increases. The Company adopted this standard on January 1, 2023, and
the adoption of ASU 2016-13 did not have a material impact on its consolidated financial statements.
|
| X |
- DefinitionLong Term Prepaid Contracts [Policy Text Block]
| Name: |
HRGN_LongTermPrepaidContractsPolicyTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionSelling And Marketing Expenses [Policy Text Block]
| Name: |
HRGN_SellingAndMarketingExpensesPolicyTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionEntity's cash and cash equivalents accounting policy with respect to restricted balances. Restrictions may include legally restricted deposits held as compensating balances against short-term borrowing arrangements, contracts entered into with others, or company statements of intention with regard to particular deposits; however, time deposits and short-term certificates of deposit are not generally included in legally restricted deposits. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(1)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-1
| Name: |
us-gaap_CashAndCashEquivalentsRestrictedCashAndCashEquivalentsPolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for credit risk. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 942
-SubTopic 825
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480981/942-825-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
| Name: |
us-gaap_ConcentrationRiskCreditRisk |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy regarding (1) the principles it follows in consolidating or combining the separate financial statements, including the principles followed in determining the inclusion or exclusion of subsidiaries or other entities in the consolidated or combined financial statements and (2) its treatment of interests (for example, common stock, a partnership interest or other means of exerting influence) in other entities, for example consolidation or use of the equity or cost methods of accounting. The accounting policy may also address the accounting treatment for intercompany accounts and transactions, noncontrolling interest, and the income statement treatment in consolidation for issuances of stock by a subsidiary. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-4
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 810
-SubTopic 10
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-1
| Name: |
us-gaap_ConsolidationPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for cost of product sold and service rendered. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Name Accounting Standards Codification
-Topic 705
-Publisher FASB
-URI https://asc.fasb.org//705/tableOfContent
| Name: |
us-gaap_CostOfSalesPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for deferral and amortization of significant deferred charges. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_DeferredChargesPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for its derivative instruments and hedging activities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 815
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480434/815-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(n))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 815
-SubTopic 10
-Section 50
-Paragraph 1A
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480434/815-10-50-1A
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 815
-SubTopic 10
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480434/815-10-50-1
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 815
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480434/815-10-50-4
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 815
-SubTopic 10
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480434/815-10-50-7
| Name: |
us-gaap_DerivativesPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for computing basic and diluted earnings or loss per share for each class of common stock and participating security. Addresses all significant policy factors, including any antidilutive items that have been excluded from the computation and takes into account stock dividends, splits and reverse splits that occur after the balance sheet date of the latest reporting period but before the issuance of the financial statements. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-2
| Name: |
us-gaap_EarningsPerSharePolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for (1) transactions denominated in a currency other than the reporting enterprise's functional currency, (2) translating foreign currency financial statements that are incorporated into the financial statements of the reporting enterprise by consolidation, combination, or the equity method of accounting, and (3) remeasurement of the financial statements of a foreign reporting enterprise in a hyperinflationary economy. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//830/tableOfContent
| Name: |
us-gaap_ForeignCurrencyTransactionsAndTranslationsPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for recognizing and measuring the impairment of long-lived assets. An entity also may disclose its accounting policy for long-lived assets to be sold. This policy excludes goodwill and intangible assets. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 5.CC)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480091/360-10-S99-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 05
-Paragraph 4
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482338/360-10-05-4
| Name: |
us-gaap_ImpairmentOrDisposalOfLongLivedAssetsPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for income taxes, which may include its accounting policies for recognizing and measuring deferred tax assets and liabilities and related valuation allowances, recognizing investment tax credits, operating loss carryforwards, tax credit carryforwards, and other carryforwards, methodologies for determining its effective income tax rate and the characterization of interest and penalties in the financial statements. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(h)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-17
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-9
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-25
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-28
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 19
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-19
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 20
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-20
| Name: |
us-gaap_IncomeTaxPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of inventory accounting policy for inventory classes, including, but not limited to, basis for determining inventory amounts, methods by which amounts are added and removed from inventory classes, loss recognition on impairment of inventories, and situations in which inventories are stated above cost. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483489/210-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(6)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 3: http://www.xbrl.org/2003/role/exampleRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 912
-SubTopic 330
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482105/912-330-50-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 330
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//330/tableOfContent
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 330
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483080/330-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 330
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483080/330-10-50-4
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 6
-Subparagraph (a)
-SubTopic 10
-Topic 270
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482989/270-10-45-6
| Name: |
us-gaap_InventoryPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy pertaining to new accounting pronouncements that may impact the entity's financial reporting. Includes, but is not limited to, quantification of the expected or actual impact.
| Name: |
us-gaap_NewAccountingPronouncementsPolicyPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for long-lived, physical asset used in normal conduct of business and not intended for resale. Includes, but is not limited to, work of art, historical treasure, and similar asset classified as collections. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-6
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for costs it has incurred (1) in a planned search or critical investigation aimed at discovery of new knowledge with the hope that such knowledge will be useful in developing a new product or service, a new process or technique, or in bringing about a significant improvement to an existing product or process; or (2) to translate research findings or other knowledge into a plan or design for a new product or process or for a significant improvement to an existing product or process. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 730
-SubTopic 10
-Name Accounting Standards Codification
-Section 05
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483044/730-10-05-1
| Name: |
us-gaap_ResearchAndDevelopmentExpensePolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for revenue. Includes revenue from contract with customer and from other sources. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-4
Reference 3: http://www.xbrl.org/2003/role/exampleRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (e)
-SubTopic 10
-Topic 235
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-4
| Name: |
us-gaap_RevenueRecognitionPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for segment reporting. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 47
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482785/280-10-55-47
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (e)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
| Name: |
us-gaap_SegmentReportingPolicyPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for inclusion of significant items in the selling, general and administrative (or similar) expense report caption. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-SubTopic 35
-Topic 720
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483406/720-35-50-1
| Name: |
us-gaap_SellingGeneralAndAdministrativeExpensesPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for award under share-based payment arrangement. Includes, but is not limited to, methodology and assumption used in measuring cost. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.C.Q3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.1.Q5)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.3.Q2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.2.Q6)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//718/tableOfContent
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationOptionAndIncentivePlansPolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for accounts receivable. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481962/310-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 310
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481569/310-20-50-1
Reference 3: http://fasb.org/us-gaap/role/ref/otherTransitionRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 11B
-Subparagraph (b)
-SubTopic 10
-Topic 310
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481962/310-10-50-11B
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481962/310-10-50-1
Reference 5: http://fasb.org/us-gaap/role/ref/otherTransitionRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-SubTopic 10
-Topic 310
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481962/310-10-50-6
Reference 6: http://fasb.org/us-gaap/role/ref/otherTransitionRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 15
-Subparagraph (d)
-SubTopic 10
-Topic 310
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481962/310-10-50-15
| Name: |
us-gaap_TradeAndOtherAccountsReceivablePolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for the use of estimates in the preparation of financial statements in conformity with generally accepted accounting principles. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-9
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-4
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 12
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-12
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-8
| Name: |
us-gaap_UseOfEstimates |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Summary of Significant Accounting Policies (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Schedule of Property Plant and Equipment Estimated Useful Lives |
Property,
plant and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets
as follows:
Schedule of Property Plant and Equipment Estimated Useful Lives
| Leasehold improvements | |
Shorter of expected useful life
or lease term |
| Computer equipment and software | |
3 years |
| Furniture, machinery and equipment | |
5-7 years |
|
| X |
- DefinitionSchedule of Property Plant And Equipment Useful Lives [Table Text Block]
| Name: |
HRGN_ScheduleOfPropertyPlantAndEquipmentUsefulLivesTableTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosurePrepaidExpensesAndOtherCurrentAssetsAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the amounts paid in advance for capitalized costs that will be expensed with the passage of time or the occurrence of a triggering event, and will be charged against earnings within one year or the normal operating cycle, if longer; the aggregate carrying amount of current assets, not separately presented elsewhere in the balance sheet; and other deferred costs.
| Name: |
us-gaap_DeferredCostsCapitalizedPrepaidAndOtherAssetsDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Property, Plant and Equipment, Net (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Property, Plant and Equipment [Abstract] |
|
| Schedule of Property Plant and Equipment Net |
Property,
plant and equipment, net consist of the following:
Schedule
of Property Plant and Equipment Net
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Leasehold improvements | |
$ | 35 | | |
$ | 35 | |
| Furniture, machinery and equipment | |
| 1,414 | | |
| 1,405 | |
| Computer equipment and software | |
| 38 | | |
| 36 | |
| Total property, plant and equipment | |
| 1,487 | | |
| 1,476 | |
| Less: accumulated depreciation | |
| (1,462 | ) | |
| (1,427 | ) |
| Property, plant and equipment, net | |
$ | 25 | | |
$ | 49 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_PropertyPlantAndEquipmentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of physical assets used in the normal conduct of business and not intended for resale. Includes, but is not limited to, balances by class of assets, depreciation and depletion expense and method used, including composite depreciation, and accumulated deprecation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Accrued and Other Current Liabilities (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Payables and Accruals [Abstract] |
|
| Schedule of Accrued and Other Current Liabilities |
Accrued
and other current liabilities consist of the following:
Schedule
of Accrued and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Advisory costs | |
$ | 325 | | |
$ | 300 | |
| Legal costs | |
| — | | |
| 135 | |
| Audit services | |
| 70 | | |
| 80 | |
| Payroll | |
| 79 | | |
| 55 | |
| Other liabilities | |
| 1 | | |
| 12 | |
| Total expenses | |
$ | 475 | | |
$ | 582 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the (a) carrying value as of the balance sheet date of liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received that are used in an entity's business (accounts payable); (b) other payables; and (c) accrued liabilities. Examples include taxes, interest, rent and utilities. Used to reflect the current portion of the liabilities (due within one year or within the normal operating cycle if longer). An alternative caption includes accrued expenses.
| Name: |
us-gaap_ScheduleOfAccountsPayableAndAccruedLiabilitiesTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Warrant Liability (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Warrant Liability |
|
| Schedule of Warrant Liability |
Schedule
of Warrant Liability
| |
|
Warrant
Liability |
|
| |
|
(in
thousands) |
|
| Balance
as of January 1, 2022 |
|
$ |
2 |
|
| Change
in fair value upon re-measurement |
|
|
(2 |
) |
| Balance
as of December 31, 2022 |
|
$ |
— |
|
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureWarrantLiabilityAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of financial instrument classified as a derivative asset (liability) after deduction of derivative liability (asset) using recurring unobservable inputs that reflect the entity's own assumption about the assumptions market participants would use in pricing. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)
-SubTopic 10
-Topic 820
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482106/820-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (b)
-SubTopic 10
-Topic 820
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482106/820-10-50-3
| Name: |
us-gaap_FairValueNetDerivativeAssetLiabilityMeasuredOnRecurringBasisUnobservableInputReconciliationTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Leases (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Leases |
|
| Schedule of Operating Leases in Consolidated Balance Sheets |
All
of the Company’s leases qualify as operating leases. The following table summarizes the presentation of the Company’s operating
leases in its consolidated balance sheets:
Schedule of Operating Leases in Consolidated Balance Sheets
| | |
| |
December 31, | |
| | |
Balance Sheet Classification | |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Assets: | |
| |
| | |
| |
| Operating lease assets | |
Right-of-use asset, net | |
$ | 48 | | |
$ | 147 | |
| Liabilities: | |
| |
| | | |
| | |
| Current portion of operating lease liabilities | |
Current portion of operating lease liabilities | |
| 48 | | |
| 99 | |
| Operating lease liabilities, net of current portion | |
Operating lease liabilities, net of current portion | |
| — | | |
| 48 | |
| Total operating lease liabilities | |
| |
$ | 48 | | |
$ | 147 | |
|
| Schedule of Weighted Average Lease Term and Discount Rates |
The
weighted average remaining lease terms and weighted average discount rates as of December 31, 2023 and 2022 were as follows:
Schedule
of Weighted Average Lease Term and Discount Rates
| | |
Year ended December 31, | |
| | |
2023 | | |
2022 | |
| Remaining lease term (in years) | |
| 0.48 | | |
| 1.43 | |
| Discount rate | |
| 14.66 | % | |
| 14.74 | % |
|
| Schedule of Operating Lease Expense Categories in Consolidated Statements of Operations |
The
following table summarizes the effect of lease costs in the Company’s consolidated statements of operations:
Schedule
of Operating Lease Expense Categories in Consolidated Statements of Operations
| | |
| |
For the Year Ended December 31, | |
| | |
| |
2023 | | |
2022 | |
| | |
| |
(in thousands) | |
| Operating lease expense | |
Research and development | |
$ | 68 | | |
$ | 77 | |
| | |
Sales and marketing | |
| 15 | | |
| — | |
| | |
General and administrative | |
| 44 | | |
| 44 | |
| | |
Total | |
$ | 127 | | |
$ | 121 | |
|
| Schedule of Minimum Lease Payments |
The
minimum lease payments for the next year is as follows:
Schedule
of Minimum Lease Payments
| | |
As of | |
| | |
December 31, 2023 | |
| | |
(in thousands) | |
| 2024 | |
$ | 50 | |
| Total lease payments | |
| 50 | |
| Less: imputed interest | |
| (2 | ) |
| Present value of operating lease liabilities | |
$ | 48 | |
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLeasesAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTable depicting details of the lessees right to asset utilization under operating lease corresponding to the relevant liability undiscounted.
| Name: |
HRGN_RightToUseOfAssetAndLiabilityInRespectOfOperatingLeasesTableTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionSchedule Of Discount Rate And Lease Term Used In Calculating Lease Liabilities And Assets [table Text Block]
| Name: |
HRGN_ScheduleOfDiscountRateAndLeaseTermUsedInCalculatingLeaseLiabilitiesAndAssetsTableTextBlock |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of lessee's lease cost. Includes, but is not limited to, interest expense for finance lease, amortization of right-of-use asset for finance lease, operating lease cost, short-term lease cost, variable lease cost and sublease income. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_LeaseCostTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of undiscounted cash flows of lessee's operating lease liability. Includes, but is not limited to, reconciliation of undiscounted cash flows to operating lease liability recognized in statement of financial position. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityMaturityTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Income Taxes (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Income Tax Disclosure [Abstract] |
|
| Schedule of Effective Income Tax |
Schedule of Effective Income Tax
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| Computed “expected” income tax benefit | |
| 21.0 | % | |
| 21.0 | % |
| State income tax benefit, net of federal income tax benefit | |
| 6.3 | % | |
| 6.3 | % |
| Tax credits | |
| 0.6 | % | |
| 0.8 | % |
| Change in valuation allowance | |
| (27.9 | )% | |
| (28.1 | )% |
| Total income taxes | |
| — | % | |
| — | % |
|
| Schedule of Deferred tax Assets and Liabilities |
The
components of the Company’s deferred tax assets and liabilities are as follows:
Schedule
of Deferred tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
Years ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Deferred tax assets: | |
| | | |
| | |
| Operating loss and credit carryforwards | |
$ | 19,167 | | |
$ | 20,487 | |
| Capitalized research and development | |
| 1,288 | | |
| 1,083 | |
| Stock-based compensation | |
| 2,504 | | |
| 1,566 | |
| Lease liabilities | |
| 13 | | |
| 40 | |
| Total deferred tax assets | |
| 22,972 | | |
| 23,176 | |
| Less: valuation allowance | |
| (22,959 | ) | |
| (23,136 | ) |
| Deferred tax assets | |
| 13 | | |
| 40 | |
| | |
| | | |
| | |
| Deferred tax liability: | |
| | | |
| | |
| Operating lease assets | |
| (13 | ) | |
| (40 | ) |
| Total deferred tax liability | |
| (13 | ) | |
| (40 | ) |
| Deferred
Tax Net | |
$ | — | | |
$ | — | |
|
| X |
- DefinitionTabular disclosure of the components of net deferred tax asset or liability recognized in an entity's statement of financial position, including the following: the total of all deferred tax liabilities, the total of all deferred tax assets, the total valuation allowance recognized for deferred tax assets. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Paragraph 2
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_ScheduleOfDeferredTaxAssetsAndLiabilitiesTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the reconciliation using percentage or dollar amounts of the reported amount of income tax expense attributable to continuing operations for the year to the amount of income tax expense that would result from applying domestic federal statutory tax rates to pretax income from continuing operations. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Paragraph 12
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-12
| Name: |
us-gaap_ScheduleOfEffectiveIncomeTaxRateReconciliationTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Series E Convertible Preferred Stock (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Series E Convertible Preferred Stock |
|
| Schedule of Categories of Preferred Stock |
Schedule
of Categories of Preferred Stock
| | |
Authorized | |
| Undesignated Preferred Stock | |
| 979,000 | |
| Series B Convertible Preferred Stock | |
| 1,000,000 | |
| Series C Convertible Preferred Stock | |
| 4,000 | |
| Series D Convertible Preferred Stock | |
| 12,000 | |
| Series E Convertible Preferred Stock | |
| 5,000 | |
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureSeriesEConvertiblePreferredStockAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of an entity's stock, including par or stated value per share, number and dollar amount of share subscriptions, shares authorized, shares issued, shares outstanding, number and dollar amount of shares held in an employee trust, dividend per share, total dividends, share conversion features, par value plus additional paid in capital, the value of treasury stock and other information necessary to a fair presentation, and EPS information. Stock by class includes common, convertible, and preferred stocks which are not redeemable or redeemable solely at the option of the issuer. Includes preferred stock with redemption features that are solely within the control of the issuer and mandatorily redeemable stock if redemption is required to occur only upon liquidation or termination of the reporting entity. If more than one issue is outstanding, state the title of each issue and the corresponding dollar amount; dollar amount of any shares subscribed but unissued and the deduction of subscriptions receivable there from; number of shares authorized, issued, and outstanding. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(27))
-SubTopic 10
-Topic 210
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-4
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-5
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481142/505-10-45-2
Reference 9: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-10
Reference 10: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-8
Reference 11: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 480
-SubTopic 10
-Section S99
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480244/480-10-S99-1
Reference 12: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-6
Reference 13: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-7
| Name: |
us-gaap_ScheduleOfStockByClassTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Common Stock (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Equity [Abstract] |
|
| Schedule of Black-Scholes Model Based on Weighted Average Assumptions |
Schedule
of Black-Scholes Model Based on Weighted Average Assumptions
| Risk-free interest rate | |
| 2.81 | % |
| Expected volatility | |
| 127.36 | % |
| Expected term | |
| 5 years | |
| Expected dividend yield | |
| — | |
| Exercise price | |
$ | 8.88 | |
| Market value of common stock | |
$ | 5.50 | |
|
| Schedule of Warrant to Purchase Common Stock |
Warrant
to purchase common stock activity for the year ended December 31, 2022 was as follows:
Schedule of Warrant to Purchase Common Stock
| | |
| | |
Weighted-average | |
| | |
Amount | | |
exercise price | |
| Outstanding at January 1, 2022 | |
| 2,501,419 | | |
$ | 4.35 | |
| Issued | |
| 427,390 | | |
| 8.88 | |
| Exercised | |
| (775,000 | ) | |
| 7.17 | |
| Expired | |
| (1,040,187 | ) | |
| 7.59 | |
| Outstanding at December 31, 2022 | |
| 1,113,622 | | |
| 4.69 | |
| Outstanding at December 31, 2023 | |
| 1,113,622 | | |
| 4.69 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_EquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the change in common stock outstanding.
| Name: |
us-gaap_ScheduleOfCommonStockOutstandingRollForwardTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of warrants or rights issued. Warrants and rights outstanding are derivative securities that give the holder the right to purchase securities (usually equity) from the issuer at a specific price within a certain time frame. Warrants are often included in a new debt issue to entice investors by a higher return potential. The main difference between warrants and call options is that warrants are issued and guaranteed by the company, whereas options are exchange instruments and are not issued by the company. Also, the lifetime of a warrant is often measured in years, while the lifetime of a typical option is measured in months. Disclose the title of issue of securities called for by warrants and rights outstanding, the aggregate amount of securities called for by warrants and rights outstanding, the date from which the warrants or rights are exercisable, and the price at which the warrant or right is exercisable. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-1
| Name: |
us-gaap_ScheduleOfStockholdersEquityNoteWarrantsOrRightsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Share-based Compensation (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Share-Based Payment Arrangement [Abstract] |
|
| Schedule of Stock Option Activity |
Stock
option activity under the Plan for the years ended December 31, 2022 and 2023 was as follows:
Schedule of Stock Option Activity
| | |
Amount | | |
Weighted-average exercise price | | |
Weighted-average contractual life (years) | | |
Aggregate intrinsic value (in thousands) | |
| Outstanding at January 1, 2022 | |
| 2,332,603 | | |
$ | 3.93 | | |
| 8.30 | | |
$ | 294 | |
| Granted | |
| 334,418 | | |
| 4.84 | | |
| | | |
| | |
| Canceled / forfeited | |
| (150,097 | ) | |
| 3.39 | | |
| | | |
| | |
| Outstanding at December 31, 2022 | |
| 2,516,924 | | |
| 3.95 | | |
| 7.68 | | |
| 6,917 | |
| Granted | |
| 2,130,007 | | |
| 5.81 | | |
| | | |
| | |
| Exercised | |
| (65,264 | ) | |
| 2.29 | | |
| | | |
| | |
| Canceled / forfeited | |
| (604,378 | ) | |
| 6.13 | | |
| | | |
| | |
| Outstanding at December 31, 2023 | |
| 3,977,289 | | |
$ | 4.64 | | |
| 7.7 | | |
$ | 5,728 | |
| Options exercisable at December 31, 2023 | |
| 2,281,655 | | |
$ | 4.64 | | |
| 7.24 | | |
$ | 4,209 | |
| Options vested or expected to vest at December 31, 2023 | |
| 3,877,871 | | |
$ | 4.69 | | |
| 7.7 | | |
$ | 5,522 | |
|
| Schedule of Weighted Average Assumptions |
The
weighted average assumptions for valuing the Company’s stock options granted were as follows:
Schedule of Weighted Average Assumptions
| | |
Year Ended December 31, | |
| | |
2023 | | |
2022 | |
| Risk-free interest rate | |
| 3.82 | % | |
| 2.74 | % |
| Expected volatility | |
| 125.35 | % | |
| 123.61 | % |
| Expected term (in years) | |
| 5.8 years | | |
| 5.8 years | |
| Expected dividend yield | |
| — | % | |
| — | % |
|
| Schedule of Share-based Compensation Expense |
Share-based
compensation expense related to the Plan for the years ended December 31, 2023 and 2022 was allocated as follows:
Schedule
of Share-based Compensation Expense
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands) | |
| Research and development | |
$ | 327 | | |
$ | 284 | |
| Selling, general and administrative | |
| 3,134 | | |
| 747 | |
| Total stock-based compensation | |
$ | 3,461 | | |
$ | 1,031 | |
|
| X |
- DefinitionTabular disclosure of allocation of amount expensed and capitalized for award under share-based payment arrangement to statement of income or comprehensive income and statement of financial position. Includes, but is not limited to, corresponding line item in financial statement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Subparagraph (h)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ScheduleOfEmployeeServiceShareBasedCompensationAllocationOfRecognizedPeriodCostsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of activity for award under share-based payment arrangement. Includes, but is not limited to, outstanding award at beginning and end of year, granted, exercised, forfeited, and weighted-average grant date fair value. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Subparagraph (c)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ScheduleOfShareBasedCompensationActivityTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the significant assumptions used during the year to estimate the fair value of stock options, including, but not limited to: (a) expected term of share options and similar instruments, (b) expected volatility of the entity's shares, (c) expected dividends, (d) risk-free rate(s), and (e) discount for post-vesting restrictions. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 718
-SubTopic 10
-Subparagraph (f)(2)
-Name Accounting Standards Codification
-Paragraph 2
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ScheduleOfShareBasedPaymentAwardStockOptionsValuationAssumptionsTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Net Loss per Share (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Earnings Per Share [Abstract] |
|
| Schedule of Basic and Diluted Net Loss Per Share |
Basic
and diluted net loss per share was calculated as follows:
Schedule of Basic and Diluted Net Loss Per Share
| | |
2023 | | |
2022 | |
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| | |
(in thousands, except shares and per share data) | |
| Net loss | |
$ | (8,945 | ) | |
$ | (6,073 | ) |
| Preferred stock dividends | |
| (77 | ) | |
| (180 | ) |
| Net loss attributable to common stockholders | |
| (9,022 | ) | |
| (6,253 | ) |
| Basic and diluted weighted average common shares outstanding | |
| 13,455,666 | | |
| 11,349,610 | |
| Basic and diluted net loss per share attributable to common stockholders | |
$ | (0.67 | ) | |
$ | (0.55 | ) |
|
| Schedule of Antidilutive Securities Excluded from Computation of Earnings per Share |
The
following potential common shares were excluded from the calculation of diluted net loss per share attributable to common stockholders
for the years ended December 31, 2023 and 2022 because including them would have had an anti-dilutive effect:
Schedule
of Antidilutive Securities Excluded from Computation of Earnings per Share
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| Warrants to purchase common stock | |
| 1,113,622 | | |
| 1,113,622 | |
| Options to purchase common stock | |
| 3,977,289 | | |
| 2,516,924 | |
| Series E convertible preferred stock | |
| — | | |
| 619,259 | |
| Total | |
| 5,090,911 | | |
| 4,249,805 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_EarningsPerShareAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of securities (including those issuable pursuant to contingent stock agreements) that could potentially dilute basic earnings per share (EPS) in the future that were not included in the computation of diluted EPS because to do so would increase EPS amounts or decrease loss per share amounts for the period presented, by antidilutive securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
| Name: |
us-gaap_ScheduleOfAntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of an entity's basic and diluted earnings per share calculations, including a reconciliation of numerators and denominators of the basic and diluted per-share computations for income from continuing operations. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
| Name: |
us-gaap_ScheduleOfEarningsPerShareBasicAndDilutedTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Segments and Geographical Information (Tables)
|
12 Months Ended |
Dec. 31, 2023 |
|---|
| Segment Reporting [Abstract] |
|
| Schedule of Reportable Segments |
The Company has two operating and reportable segments: i) Regenerative Biotech
focused on the development of regenerative medicine treatments with operations currently in the United States and ii) Longevity Products
relating to longevity products with operations currently in Asia. The following table presents the Company’s reportable
segment results for the year ended 2023:
Schedule of Reportable Segments
| | |
| | | |
| | |
|
| | |
| | |
Regenerative Biotech | | |
Longevity Products | |
|
Total | |
| 2023 | |
| | | |
| | |
|
| | |
| Revenues | |
$ | — | | |
$ | 103 | |
|
$ | 103 | |
| Net loss | |
| (8,677 | ) | |
| (268 | ) |
|
| (8,945 | ) |
| Total assets | |
| 2,426 | | |
| 188 | |
|
| 2,614 | |
|
| X |
- DefinitionTabular disclosure of the profit or loss and total assets for each reportable segment. An entity discloses certain information on each reportable segment if the amounts (a) are included in the measure of segment profit or loss reviewed by the chief operating decision maker or (b) are otherwise regularly provided to the chief operating decision maker, even if not included in that measure of segment profit or loss. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 350
-SubTopic 20
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482573/350-20-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 280
-SubTopic 10
-Section 50
-Paragraph 25
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-25
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 280
-SubTopic 10
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 280
-SubTopic 10
-Section 50
-Paragraph 30
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
| Name: |
us-gaap_ScheduleOfSegmentReportingInformationBySegmentTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_SegmentReportingAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
| X |
- DefinitionCash includes currency on hand as well as demand deposits with banks or financial institutions. It also includes other kinds of accounts that have the general characteristics of demand deposits in that the customer may deposit additional funds at any time and effectively may withdraw funds at any time without prior notice or penalty. Cash equivalents, excluding items classified as marketable securities, include short-term, highly liquid Investments that are both readily convertible to known amounts of cash, and so near their maturity that they present minimal risk of changes in value because of changes in interest rates. Generally, only investments with original maturities of three months or less qualify under that definition. Original maturity means original maturity to the entity holding the investment. For example, both a three-month US Treasury bill and a three-year Treasury note purchased three months from maturity qualify as cash equivalents. However, a Treasury note purchased three years ago does not become a cash equivalent when its remaining maturity is three months. Short-term investments, exclusive of cash equivalents, generally consist of marketable securities intended to be sold within one year (or the normal operating cycle if longer) and may include trading securities, available-for-sale securities, or held-to-maturity securities (if maturing within one year), as applicable. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CashCashEquivalentsAndShortTermInvestments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe cash inflow from bank borrowing during the year. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 14
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromBankDebt |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from parent as a source of financing that is recorded as additional paid in capital. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 14
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromContributionsFromParent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of accumulated undistributed earnings (deficit). Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (g)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (h)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480990/946-20-50-11
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(23)(a)(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30)(a)(3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_RetainedEarningsAccumulatedDeficit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_PlanNameAxis=HRGN_HarvardBiosciencePlanMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Schedule of Property Plant and Equipment Estimated Useful Lives (Details)
|
Dec. 31, 2023 |
| Computer Equipment and Software [Member] |
|
| Property, Plant and Equipment [Line Items] |
|
| Property, plant and equipment, useful life |
3 years
|
| Furniture Machinery And Equipment [Member] | Minimum [Member] |
|
| Property, Plant and Equipment [Line Items] |
|
| Property, plant and equipment, useful life |
5 years
|
| Furniture Machinery And Equipment [Member] | Maximum [Member] |
|
| Property, Plant and Equipment [Line Items] |
|
| Property, plant and equipment, useful life |
7 years
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_PropertyPlantAndEquipmentLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionUseful life of long lived, physical assets used in the normal conduct of business and not intended for resale, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents the reported fact of one year, five months, and thirteen days. Examples include, but not limited to, land, buildings, machinery and equipment, office equipment, furniture and fixtures, and computer equipment.
| Name: |
us-gaap_PropertyPlantAndEquipmentUsefulLife |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
us-gaap_PropertyPlantAndEquipmentByTypeAxis=HRGN_ComputerEquipmentAndSoftwareMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_PropertyPlantAndEquipmentByTypeAxis=HRGN_FurnitureMachineryAndEquipmentMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_CashAndCashEquivalentsLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Excludes cash and cash equivalents within disposal group and discontinued operation. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(2))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CashEquivalentsAtCarryingValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe amount of cash deposited in financial institutions as of the balance sheet date that is insured by the Federal Deposit Insurance Corporation.
| Name: |
us-gaap_CashFDICInsuredAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe aggregate amount of write-downs for impairments recognized during the period for long lived assets held for use (including those held for disposal by means other than sale). Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 360
-SubTopic 10
-Section 45
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482130/360-10-45-4
| Name: |
us-gaap_ImpairmentOfLongLivedAssetsHeldForUse |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of cash paid for deposits on goods and services during the period; excludes time deposits and deposits with other institutions, which pertain to financial service entities. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
| Name: |
us-gaap_PaymentsForDeposits |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionPeriod over which grantee's right to exercise award under share-based payment arrangement is no longer contingent on satisfaction of service or performance condition, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Includes, but is not limited to, combination of market, performance or service condition. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardAwardVestingPeriod1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cumulative translation gain (loss), after tax, from translating foreign currency financial statements into the reporting currency. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-11
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
| Name: |
us-gaap_TranslationAdjustmentFunctionalToReportingCurrencyNetOfTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=us-gaap_RestrictedStockUnitsRSUMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_CashAndCashEquivalentsAxis=us-gaap_MoneyMarketFundsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionFair value transfers between level hierarchies.
| Name: |
HRGN_FairValueTransfersBetweenLevelHierarchies |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_FairValueDisclosuresAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of investments including trading securities, available-for-sale securities, held-to-maturity securities, and short-term investments classified as other and current. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_ShortTermInvestments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosurePrepaidExpensesAndOtherCurrentAssetsAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPrepaid expense contracts.
| Name: |
HRGN_PrepaidExpenseContracts |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of asset related to consideration paid in advance for costs that provide economic benefits in future periods, and amount of other assets that are expected to be realized or consumed within one year or the normal operating cycle, if longer. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PrepaidExpenseAndOtherAssetsCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of asset related to consideration paid in advance for insurance that provides economic benefits within a future period of one year or the normal operating cycle, if longer. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (g)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-1
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 340
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483032/340-10-45-1
Reference 3: http://www.xbrl.org/2003/role/exampleRef
-Topic 340
-SubTopic 10
-Name Accounting Standards Codification
-Section 05
-Paragraph 5
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482955/340-10-05-5
| Name: |
us-gaap_PrepaidInsurance |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
v3.24.1
Schedule of Property Plant and Equipment Net (Details) - USD ($)
$ in Thousands |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Property, Plant and Equipment [Line Items] |
|
|
| Total property, plant and equipment |
$ 1,487
|
$ 1,476
|
| Less: accumulated depreciation |
(1,462)
|
(1,427)
|
| Property, plant and equipment, net |
25
|
49
|
| Leasehold Improvements [Member] |
|
|
| Property, Plant and Equipment [Line Items] |
|
|
| Total property, plant and equipment |
35
|
35
|
| Machinery and Equipment [Member] |
|
|
| Property, Plant and Equipment [Line Items] |
|
|
| Total property, plant and equipment |
1,414
|
1,405
|
| Computer Equipment [Member] |
|
|
| Property, Plant and Equipment [Line Items] |
|
|
| Total property, plant and equipment |
$ 38
|
$ 36
|
| X |
- DefinitionAmount of accumulated depreciation, depletion and amortization for physical assets used in the normal conduct of business to produce goods and services. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(14))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 360
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_AccumulatedDepreciationDepletionAndAmortizationPropertyPlantAndEquipment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before accumulated depreciation, depletion and amortization of physical assets used in the normal conduct of business and not intended for resale. Examples include, but are not limited to, land, buildings, machinery and equipment, office equipment, and furniture and fixtures. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(13))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_PropertyPlantAndEquipmentLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount after accumulated depreciation, depletion and amortization of physical assets used in the normal conduct of business to produce goods and services and not intended for resale. Examples include, but are not limited to, land, buildings, machinery and equipment, office equipment, and furniture and fixtures. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 360
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480842/942-360-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
us-gaap_PropertyPlantAndEquipmentByTypeAxis=us-gaap_LeaseholdImprovementsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_PropertyPlantAndEquipmentByTypeAxis=us-gaap_MachineryAndEquipmentMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_PropertyPlantAndEquipmentByTypeAxis=us-gaap_ComputerEquipmentMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionThe amount of expense recognized in the current period that reflects the allocation of the cost of tangible assets over the assets' useful lives. Includes production and non-production related depreciation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 360
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_Depreciation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_PropertyPlantAndEquipmentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLongtermPrepaidContractsAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of cash paid for deposits on goods and services during the period; excludes time deposits and deposits with other institutions, which pertain to financial service entities. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
| Name: |
us-gaap_PaymentsForDeposits |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
v3.24.1
| X |
- DefinitionAccrued advisory costs current.
| Name: |
HRGN_AccruedAdvisoryCostsCurrent |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAccrued audit services current.
| Name: |
HRGN_AccruedAuditServicesCurrent |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAccrued legal expenses current.
| Name: |
HRGN_AccruedLegalExpensesCurrent |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying values as of the balance sheet date of obligations incurred through that date and due within one year (or the operating cycle, if longer), including liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received, taxes, interest, rent and utilities, accrued salaries and bonuses, payroll taxes and fringe benefits. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19,20)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AccountsPayableAndAccruedLiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of liabilities classified as other, due within one year or the normal operating cycle, if longer. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.20)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_OtherLiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosureWarrantLiabilityAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Warrant Liability (Details Narrative) - shares
|
Dec. 31, 2023 |
Feb. 28, 2022 |
Dec. 31, 2017 |
| Warrant Liability |
|
|
|
| Warrants to be re-measured, number |
92,212
|
|
1,044,396
|
| Warrants to be modified, number |
|
|
952,184
|
| Warrants expired and unexercised |
|
92,212
|
|
| X |
- References
+ Details
| Name: |
HRGN_DisclosureWarrantLiabilityAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionShare based compensation arrangement by share based payment award warrants expired.
| Name: |
HRGN_SharebasedCompensationArrangementBySharebasedPaymentAwardWarrantsExpired |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWarrants to be modified number.
| Name: |
HRGN_WarrantsToBeModifiedNumber |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWarrants to be remeasured number.
| Name: |
HRGN_WarrantsToBeRemeasuredNumber |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
v3.24.1
Commitments and Contingencies (Details Narrative) - USD ($)
|
|
|
6 Months Ended |
12 Months Ended |
Jun. 10, 2022 |
Mar. 03, 2022 |
Jun. 30, 2022 |
Dec. 31, 2023 |
| Loss Contingencies [Line Items] |
|
|
|
|
| Estimated litigation cost |
|
|
|
$ 5,900,000
|
| Harvard Bioscience Inc. [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Litigation amount received |
|
|
$ 4,000,000.0
|
$ 4,000,000.0
|
| Harvard Bioscience Inc. [Member] | Convertible Preferred Stock [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Preferred stock, dividend rate, percentage |
|
|
|
8.00%
|
| Harvard Bioscience Inc. [Member] | Series E 8% Convertible Preferred Stock [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Litigation amount received |
$ 4,000,000.0
|
|
|
|
| Preferred stock, dividend rate, percentage |
8.00%
|
|
|
|
| Temporary equity, stock issued |
4,000
|
|
|
|
| Temporary equity, issue price per share |
$ 1,000
|
|
|
|
| Accrual for contingency matter |
$ 3,300,000
|
|
|
|
| Legal costs on claims against insurance carrier |
$ 800,000
|
|
|
|
| Yale University [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Prepaid Expense |
|
|
|
$ 130,000
|
| McGowan Institute for Regenerative Medicine [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Prepaid Expense |
|
|
|
$ 61,000
|
| Medmarc [Member] |
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
| Cash payment received on litigation |
|
$ 100,000
|
|
|
| X |
- DefinitionIssue price per share of temporary equity.
| Name: |
HRGN_TemporaryEquityIssuePricePerShare |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares classified as temporary equity issued during the period.
| Name: |
HRGN_TemporaryEquityStockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount awarded from other party in judgment or settlement of litigation.
| Name: |
us-gaap_LitigationSettlementAmountAwardedFromOtherParty |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of litigation expense, including but not limited to legal, forensic, accounting, and investigative fees.
| Name: |
us-gaap_LitigationSettlementExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 460
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482425/460-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-1
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-4
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-9
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-9
| Name: |
us-gaap_LossContingenciesLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of loss contingency liability. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-1
Reference 2: http://www.xbrl.org/2003/role/recommendedDisclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-10
| Name: |
us-gaap_LossContingencyAccrualAtCarryingValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionReflects the estimated amount of loss from the specified contingency as of the balance sheet date. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 460
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482425/460-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-4
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-9
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 460
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482425/460-10-50-2
| Name: |
us-gaap_LossContingencyEstimateOfPossibleLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of cash inflow from the collection of receivables related to a loss contingency. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 460
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482425/460-10-50-3
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-17
| Name: |
us-gaap_LossContingencyReceivableProceeds |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe percentage rate used to calculate dividend payments on preferred stock. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 320
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.12-12A(Column A)(Footnote 3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480032/946-320-S99-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 320
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.12-12(Column A)(Footnote 4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480032/946-320-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 320
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.12-12B(Column A)(Footnote 3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480032/946-320-S99-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 320
-Name Accounting Standards Codification
-Section S99
-Paragraph 6
-Subparagraph (SX 210.12-14(Column A)(Footnote 3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480032/946-320-S99-6
| Name: |
us-gaap_PreferredStockDividendRatePercentage |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCarrying amount as of the balance sheet date of expenditures made in advance of when the economic benefit of the cost will be realized, and which will be expensed in future periods with the passage of time or when a triggering event occurs. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(10))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(7)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 942
-SubTopic 210
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03.10)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_PrepaidExpenseCurrentAndNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=HRGN_HarvardBioscienceInc.Member |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_ConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=HRGN_SeriesEEightPercentageConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=HRGN_YaleUniversityMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=HRGN_McGowanInstituteForRegenerativeMedicineMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_LitigationCaseAxis=HRGN_MedmarcMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLeasesAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiability |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as current. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as noncurrent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's right to use underlying asset under operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseRightOfUseAsset |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLeasesAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average discount rate for operating lease calculated at point in time. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseWeightedAverageDiscountRatePercent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average remaining lease term for operating lease, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseWeightedAverageRemainingLeaseTerm1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
v3.24.1
| X |
- DefinitionAmount of single lease cost, calculated by allocation of remaining cost of lease over remaining lease term. Includes, but is not limited to, single lease cost, after impairment of right-of-use asset, calculated by amortization of remaining right-of-use asset and accretion of lease liability. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_ResearchAndDevelopmentExpenseMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_SellingAndMarketingExpenseMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_SellingGeneralAndAdministrativeExpensesMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- References
+ Details
| Name: |
HRGN_DisclosureLeasesAbstract |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease to be paid in next fiscal year following current fiscal year. Excludes interim and annual periods when interim periods are reported from current statement of financial position date (rolling approach). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDueNextTwelveMonths |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payments in excess of discounted obligation for lease payments for operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityUndiscountedExcessAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiability |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.24.1
| X |
- DefinitionRemaining lease term of operating lease, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-3
| Name: |
us-gaap_LesseeOperatingLeaseRemainingLeaseTerm |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of cash outflow from operating lease, excluding payments to bring another asset to condition and location necessary for its intended use. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 5
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-5
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeasePayments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
v3.24.1
Schedule of Deferred tax Assets and Liabilities (Details) - USD ($)
$ in Thousands |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Income Tax Disclosure [Abstract] |
|
|
| Operating loss and credit carryforwards |
$ 19,167
|
$ 20,487
|
| Capitalized research and development |
1,288
|
1,083
|
| Stock-based compensation |
2,504
|
1,566
|
| Lease liabilities |
13
|
40
|
| Total deferred tax assets |
22,972
|
23,176
|
| Less: valuation allowance |
(22,959)
|
(23,136)
|
| Deferred tax assets |
13
|
40
|
| Operating lease assets |
(13)
|
(40)
|
| Total deferred tax liability |
(13)
|
(40)
|
| Deferred Tax Net |
|
|
| X |
- DefinitionDeferred tax assets lease liabilities.
| Name: |
HRGN_DeferredTaxAssetsLeaseLiabilities |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDeferred tax liability operating lease assets.
| Name: |
HRGN_DeferredTaxLiabilityOperatingLeaseAssets |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences and carryforwards. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences from in-process research and development costs expensed in connection with a business combination. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsInProcessResearchAndDevelopment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount after allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences and carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible operating loss carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsOperatingLossCarryforwards |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences from share-based compensation. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsTaxDeferredExpenseCompensationAndBenefitsShareBasedCompensationCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of deferred tax assets for which it is more likely than not that a tax benefit will not be realized. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsValuationAllowance |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount, after deferred tax asset, of deferred tax liability attributable to taxable differences without jurisdictional netting. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 45
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-6
| Name: |
us-gaap_DeferredTaxLiabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.24.1
Income Taxes (Details Narrative) - USD ($)
|
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Operating Loss Carryforwards [Line Items] |
|
|
| Expected federal tax rate |
21.00%
|
21.00%
|
| Increase decrease in valuation allowance |
$ 200,000
|
$ 2,900,000
|
| Utilized to offset of taxable income percentage |
80.00%
|
|
| Unrecognized tax benefits |
$ 0
|
$ 0
|
| Domestic Tax Authority [Member] |
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
| Operating loss carryforwards |
68,700,000
|
|
| Deferred tax assets, research and development credit carryforwards |
1,500,000
|
|
| State and Local Jurisdiction [Member] |
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
| Operating loss carryforwards |
68,100,000
|
|
| Deferred tax assets, operating loss carryforwards, not subject to expiration |
42,300,000
|
|
| Deferred tax assets, research and development credit carryforwards |
$ 1,000,000.0
|
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible operating loss carryforwards that are not subject to expiration dates.
| Name: |
us-gaap_DeferredTaxAssetsOperatingLossCarryforwardsNotSubjectToExpiration |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible research tax credit carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_DeferredTaxAssetsTaxCreditCarryforwardsResearch |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of operating loss carryforward, before tax effects, available to reduce future taxable income under enacted tax laws. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_OperatingLossCarryforwards |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_OperatingLossCarryforwardsLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of unrecognized tax benefits. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 15A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-15A
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10B
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-10B
| Name: |
us-gaap_UnrecognizedTaxBenefits |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of increase (decrease) in the valuation allowance for a specified deferred tax asset. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_ValuationAllowanceDeferredTaxAssetChangeInAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
v3.24.1
| X |
- References
+ Details
| Name: |
us-gaap_CompensationAndRetirementDisclosureAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cost for defined contribution plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 715
-SubTopic 70
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480794/715-70-50-1
| Name: |
us-gaap_DefinedContributionPlanCostRecognized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
v3.24.1
Schedule of Categories of Preferred Stock (Details)
|
Dec. 31, 2023
shares
|
| Undesignated Preferred Stock [Member] |
|
| Preferred stock, shares authorized |
979,000
|
| Series B Preferred Stock [Member] |
|
| Preferred stock, shares authorized |
1,000,000
|
| Series C Preferred Stock [Member] |
|
| Preferred stock, shares authorized |
4,000
|
| Series D Preferred Stock [Member] |
|
| Preferred stock, shares authorized |
12,000
|
| Series E Preferred Stock [Member] |
|
| Preferred stock, shares authorized |
5,000
|
| X |
- DefinitionThe maximum number of nonredeemable preferred shares (or preferred stock redeemable solely at the option of the issuer) permitted to be issued by an entity's charter and bylaws. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PreferredStockSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=HRGN_UndesignatedPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_SeriesBPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_SeriesCPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_SeriesDPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_SeriesEPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Series E Convertible Preferred Stock (Details Narrative) - USD ($)
|
Apr. 12, 2023 |
Jan. 18, 2023 |
Jun. 10, 2022 |
Apr. 28, 2022 |
Dec. 31, 2023 |
| Preferred stock, shares outstanding |
|
|
|
|
0
|
| Private Placement [Member] |
|
|
|
|
|
| Proceeds from private placement |
$ 6,000,000.0
|
|
|
|
|
| Common Stock [Member] |
|
|
|
|
|
| Common stock dividends, shares |
|
31,933
|
|
|
|
| Series E Convertible Preferred Stock [Member] |
|
|
|
|
|
| Conversion of stock, shares issued |
|
200
|
|
|
|
| Accrued dividends |
|
$ 9,545
|
|
|
|
| Series E Preferred Stock [Member] |
|
|
|
|
|
| Conversion of stock, shares issued |
674,693
|
|
|
|
|
| Conversion price, per share |
$ 6.00
|
|
|
|
|
| Series E Preferred Stock [Member] | Private Placement [Member] |
|
|
|
|
|
| Proceeds from private placement |
$ 4,000,000.0
|
|
|
|
|
| Harvard Bioscience Inc [Member] | Series E Convertible Preferred Stock [Member] |
|
|
|
|
|
| Litigation amount received |
|
|
$ 4,000,000.0
|
$ 4,000,000.0
|
|
| Temporary equity, stock issued |
|
|
4,000
|
|
|
| Temporary equity, issue price per share |
|
|
$ 1,000
|
|
|
| X |
- DefinitionIssue price per share of temporary equity.
| Name: |
HRGN_TemporaryEquityIssuePricePerShare |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares classified as temporary equity issued during the period.
| Name: |
HRGN_TemporaryEquityStockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares of common stock issued as dividends during the period. Excludes stock splits. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
| Name: |
us-gaap_CommonStockDividendsShares |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of new shares issued in the conversion of stock in a noncash (or part noncash) transaction. Noncash is defined as transactions during a period that do not result in cash receipts or cash payments in the period. "Part noncash" refers to that portion of the transaction not resulting in cash receipts or cash payments in the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-4
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-3
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-5
| Name: |
us-gaap_ConversionOfStockSharesIssued1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount awarded from other party in judgment or settlement of litigation.
| Name: |
us-gaap_LitigationSettlementAmountAwardedFromOtherParty |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionPer share conversion price of preferred stock. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
| Name: |
us-gaap_PreferredStockConvertibleConversionPrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAggregate share number for all nonredeemable preferred stock (or preferred stock redeemable solely at the option of the issuer) held by stockholders. Does not include preferred shares that have been repurchased. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PreferredStockSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe cash inflow associated with the amount received from entity's raising of capital via private rather than public placement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfPrivatePlacement |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionValue of stock issued to shareholders as a dividend during the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-31)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueStockDividend |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_SubsidiarySaleOfStockAxis=us-gaap_PrivatePlacementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_CommonStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=HRGN_SeriesEConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=us-gaap_SeriesEPreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=HRGN_HarvardBioscienceIncMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionFair value assumption warrant risk free interest rate.
| Name: |
HRGN_FairValueAssumptionWarrantRiskFreeInterestRate |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFair value assumptions warrants expected dividend yield.
| Name: |
HRGN_FairValueAssumptionsWarrantsExpectedDividendYield |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFair value assumptions warrants expected terms.
| Name: |
HRGN_FairValueAssumptionsWarrantsExpectedTerms |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFair value assumptions warrants expected volatility rates.
| Name: |
HRGN_FairValueAssumptionsWarrantsExpectedVolatilityRates |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionExercise price per share or per unit of warrants or rights outstanding. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightExercisePriceOfWarrantsOrRights1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_EquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPrice of a single share of a number of saleable stocks of a company.
| Name: |
us-gaap_SharePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
v3.24.1
Schedule of Warrant to Purchase Common Stock (Details) - $ / shares
|
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
| Outstanding, balance |
2,516,924
|
2,332,603
|
| Weighted-average exercise price, Outstanding |
$ 3.95
|
$ 3.93
|
| Issued |
2,130,007
|
334,418
|
| Weighted-average exercise price, Issued |
$ 5.81
|
$ 4.84
|
| Exercised |
(65,264)
|
|
| Weighted-average exercise price, Exercised |
$ 2.29
|
|
| Outstanding, balance |
3,977,289
|
2,516,924
|
| Weighted-average exercise price, Outstanding |
$ 4.64
|
$ 3.95
|
| Warrant [Member] |
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
| Outstanding, balance |
1,113,622
|
2,501,419
|
| Weighted-average exercise price, Outstanding |
$ 4.69
|
$ 4.35
|
| Issued |
|
427,390
|
| Weighted-average exercise price, Issued |
|
$ 8.88
|
| Exercised |
|
(775,000)
|
| Weighted-average exercise price, Exercised |
|
$ 7.17
|
| Expired |
|
(1,040,187)
|
| Weighted-average exercise price, Expired |
|
$ 7.59
|
| Outstanding, balance |
1,113,622
|
1,113,622
|
| Weighted-average exercise price, Outstanding |
$ 4.69
|
$ 4.69
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-4
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481674/830-30-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-17
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
| Name: |
us-gaap_AccumulatedOtherComprehensiveIncomeLossLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of options or other stock instruments for which the right to exercise has lapsed under the terms of the plan agreements. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsExpirationsInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionGross number of share options (or share units) granted during the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsGrantsInPeriodGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of options outstanding, including both vested and non-vested options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average price at which grantees can acquire the shares reserved for issuance under the stock option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average price at which option holders acquired shares when converting their stock options into shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsExercisesInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average price at which grantees could have acquired the underlying shares with respect to stock options of the plan that expired. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsExpirationsInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average per share amount at which grantees can acquire shares of common stock by exercise of options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsGrantsInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of share options (or share units) exercised during the current period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesStockOptionsExercised |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_WarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Common Stock (Details Narrative) - USD ($)
$ / shares in Units, $ in Millions |
|
|
|
1 Months Ended |
6 Months Ended |
12 Months Ended |
Apr. 12, 2023 |
May 16, 2022 |
May 12, 2022 |
Jun. 30, 2022 |
Jun. 30, 2022 |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Common stock, shares authorized |
|
|
|
|
|
60,000,000
|
60,000,000
|
| Shares available for issuance |
|
|
|
|
|
40,961,765
|
|
| Employee Stock Purchase Plan [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Shares available for issuance |
|
|
|
|
|
2,966
|
2,966
|
| Employees purchase shares of common stock, percent |
|
|
|
|
|
85.00%
|
|
| Share-based compensation arrangement by share-based payment award, number of shares authorized |
|
|
|
|
|
7,500
|
|
| Share-based compensation arrangement by share-based payment award, shares issued in period |
|
|
|
|
|
4,534
|
|
| Harvard Bioscience Inc. [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Litigation amount received |
|
|
|
|
$ 4.0
|
$ 4.0
|
|
| Harvard Bioscience Inc. [Member] | Series E Convertible Preferred Stock [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Temporary equity, issue shares |
|
|
|
4,000
|
|
|
180
|
| Temporary equity, issue price per share |
|
|
|
$ 1,000
|
|
|
|
| Litigation amount received |
|
|
|
$ 4.0
|
|
|
|
| Private Placement [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Amount of consideration received from the transaction |
$ 6.0
|
$ 5.1
|
$ 5.1
|
|
|
|
|
| Price per share |
$ 6.00
|
|
$ 5.92
|
|
|
|
|
| Cash received on stock transaction after deduction of costs |
|
5.1
|
|
|
|
|
|
| Common Stock [Member] | Private Placement [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Number of shares issued in transaction |
1,000,967
|
|
854,771
|
|
|
|
|
| Number of securities into which the class of warrant or right may be converted |
|
|
427,390
|
|
|
|
|
| Number of securities into which each warrant or right may be converted |
|
|
0.5
|
|
|
|
|
| Cash proceeds received |
|
3.6
|
|
|
|
|
|
| Warrant [Member] | Private Placement [Member] |
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
| Cash received on stock transaction after deduction of costs |
|
$ 1.5
|
|
|
|
|
|
| X |
- DefinitionIssue price per share of temporary equity.
| Name: |
HRGN_TemporaryEquityIssuePricePerShare |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares classified as temporary equity issued during the period.
| Name: |
HRGN_TemporaryEquityStockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-4
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481674/830-30-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-17
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
| Name: |
us-gaap_AccumulatedOtherComprehensiveIncomeLossLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of securities into which each warrant or right may be converted. For example, but not limited to, each warrant may be converted into two shares.
| Name: |
us-gaap_ClassOfWarrantOrRightNumberOfSecuritiesCalledByEachWarrantOrRight |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of securities into which the class of warrant or right may be converted. For example, but not limited to, 500,000 warrants may be converted into 1,000,000 shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightNumberOfSecuritiesCalledByWarrantsOrRights |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAggregate number of common shares reserved for future issuance. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockCapitalSharesReservedForFutureIssuance |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe maximum number of common shares permitted to be issued by an entity's charter and bylaws. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAmount awarded from other party in judgment or settlement of litigation.
| Name: |
us-gaap_LitigationSettlementAmountAwardedFromOtherParty |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from the issuance of common stock, preferred stock, treasury stock, stock options, and other types of equity. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(i)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
| Name: |
us-gaap_ProceedsFromIssuanceOrSaleOfEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionCash received on stock transaction after deduction of issuance costs.
| Name: |
us-gaap_SaleOfStockConsiderationReceivedOnTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of consideration received by subsidiary or equity investee in exchange for shares of stock issued or sold. Includes amount of cash received, fair value of noncash assets received, and fair value of liabilities assumed by the investor.
| Name: |
us-gaap_SaleOfStockConsiderationReceivedPerTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of shares issued or sold by the subsidiary or equity method investee per stock transaction.
| Name: |
us-gaap_SaleOfStockNumberOfSharesIssuedInTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPer share amount received by subsidiary or equity investee for each share of common stock issued or sold in the stock transaction.
| Name: |
us-gaap_SaleOfStockPricePerShare |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares authorized for issuance under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardNumberOfSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares issued under share-based payment arrangement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardSharesIssuedInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPurchase price of common stock expressed as a percentage of its fair value.
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardPurchasePriceOfCommonStockPercent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_PlanNameAxis=HRGN_EmployeeStockPurchasePlanMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=HRGN_HarvardBioscienceInc.Member |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=HRGN_SeriesEConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_SubsidiarySaleOfStockAxis=us-gaap_PrivatePlacementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_CommonStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_WarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Schedule of Stock Option Activity (Details) - USD ($)
$ / shares in Units, $ in Thousands |
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Share-Based Payment Arrangement [Abstract] |
|
|
| Outstanding, balance |
2,516,924
|
2,332,603
|
| Weighted-average exercise price, Outstanding |
$ 3.95
|
$ 3.93
|
| Weighted-average contractual life, Outstanding |
7 years 8 months 4 days
|
8 years 3 months 18 days
|
| Aggregate intrinsic value, Outstanding (in dollars) |
$ 6,917
|
$ 294
|
| Granted |
2,130,007
|
334,418
|
| Weighted-average exercise price, Granted |
$ 5.81
|
$ 4.84
|
| Canceled / forfeited |
(604,378)
|
(150,097)
|
| Weighted-average exercise price, Canceled / forfeited |
$ 6.13
|
$ 3.39
|
| Exercised |
(65,264)
|
|
| Weighted-average exercise price, Exercised |
$ 2.29
|
|
| Outstanding, balance |
3,977,289
|
2,516,924
|
| Weighted-average exercise price, Outstanding |
$ 4.64
|
$ 3.95
|
| Weighted-average contractual life, Options vested and expected to vest |
7 years 8 months 12 days
|
|
| Aggregate intrinsic value, Outstanding (in dollars) |
$ 5,728
|
$ 6,917
|
| Options exercisable |
2,281,655
|
|
| Weighted-average exercise price, Options exercisable |
$ 4.64
|
|
| Weighted-average contractual life, Options exercisable |
7 years 2 months 26 days
|
|
| Outstanding at the beginning (in dollars) |
$ 4,209
|
|
| Options vested or expected to vest |
3,877,871
|
|
| Weighted-average exercise price, Options vested and expected to vest |
$ 4.69
|
|
| Aggregate intrinsic value, Options vested and expected to vest (in dollars) |
$ 5,522
|
|
| X |
- DefinitionThe number of shares into which fully or partially vested stock options outstanding as of the balance sheet date can be currently converted under the option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsExercisableNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe weighted-average price as of the balance sheet date at which grantees can acquire the shares reserved for issuance on vested portions of options outstanding and currently exercisable under the stock option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsExercisableWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe number of shares under options that were cancelled during the reporting period as a result of occurrence of a terminating event specified in contractual agreements pertaining to the stock option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsForfeituresInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionGross number of share options (or share units) granted during the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsGrantsInPeriodGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount by which the current fair value of the underlying stock exceeds the exercise price of options outstanding. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingIntrinsicValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of options outstanding, including both vested and non-vested options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average price at which grantees can acquire the shares reserved for issuance under the stock option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAmount by which current fair value of underlying stock exceeds exercise price of fully vested and expected to vest options outstanding. Includes, but is not limited to, unvested options for which requisite service period has not been rendered but that are expected to vest based on achievement of performance condition, if forfeitures are recognized when they occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsVestedAndExpectedToVestOutstandingAggregateIntrinsicValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of fully vested and expected to vest options outstanding that can be converted into shares under option plan. Includes, but is not limited to, unvested options for which requisite service period has not been rendered but that are expected to vest based on achievement of performance condition, if forfeitures are recognized when they occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsVestedAndExpectedToVestOutstandingNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted-average exercise price, at which grantee can acquire shares reserved for issuance, for fully vested and expected to vest options outstanding. Includes, but is not limited to, unvested options for which requisite service period has not been rendered but that are expected to vest based on achievement of performance condition, if forfeitures are recognized when they occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsVestedAndExpectedToVestOutstandingWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average price at which option holders acquired shares when converting their stock options into shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsExercisesInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average price at which grantees could have acquired the underlying shares with respect to stock options that were terminated. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsForfeituresInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average per share amount at which grantees can acquire shares of common stock by exercise of options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementsByShareBasedPaymentAwardOptionsGrantsInPeriodWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of difference between fair value of the underlying shares reserved for issuance and exercise price of vested portions of options outstanding and currently exercisable. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardOptionsExercisableIntrinsicValue1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average remaining contractual term for vested portions of options outstanding and currently exercisable or convertible, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents the reported fact of one year, five months, and thirteen days. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardOptionsExercisableWeightedAverageRemainingContractualTerm1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average remaining contractual term for option awards outstanding, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents the reported fact of one year, five months, and thirteen days. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 718
-SubTopic 10
-Subparagraph (e)(1)
-Name Accounting Standards Codification
-Paragraph 2
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardOptionsOutstandingWeightedAverageRemainingContractualTerm2 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average remaining contractual term for fully vested and expected to vest exercisable or convertible options, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents the reported fact of one year, five months, and thirteen days. Includes, but is not limited to, unvested options for which requisite service period has not been rendered but that are expected to vest based on achievement of performance condition, if forfeitures are recognized when they occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardOptionsVestedAndExpectedToVestExercisableWeightedAverageRemainingContractualTerm1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of share options (or share units) exercised during the current period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesStockOptionsExercised |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
| X |
- DefinitionThe estimated dividend rate (a percentage of the share price) to be paid (expected dividends) to holders of the underlying shares over the option's term. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardFairValueAssumptionsExpectedDividendRate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe estimated measure of the percentage by which a share price is expected to fluctuate during a period. Volatility also may be defined as a probability-weighted measure of the dispersion of returns about the mean. The volatility of a share price is the standard deviation of the continuously compounded rates of return on the share over a specified period. That is the same as the standard deviation of the differences in the natural logarithms of the stock prices plus dividends, if any, over the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardFairValueAssumptionsExpectedVolatilityRate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe risk-free interest rate assumption that is used in valuing an option on its own shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardFairValueAssumptionsRiskFreeInterestRate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionExpected term of award under share-based payment arrangement, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardFairValueAssumptionsExpectedTerm1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Schedule of Share-based Compensation Expense (Details) - Biostage 2013 Equity Incentive Plan [Member] - USD ($)
$ in Thousands |
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Share-Based Payment Arrangement, Expensed and Capitalized, Amount [Line Items] |
|
|
| Total share-based compensation |
$ 3,461
|
$ 1,031
|
| Research and Development Expense [Member] |
|
|
| Share-Based Payment Arrangement, Expensed and Capitalized, Amount [Line Items] |
|
|
| Total share-based compensation |
327
|
284
|
| Selling, General and Administrative Expenses [Member] |
|
|
| Share-Based Payment Arrangement, Expensed and Capitalized, Amount [Line Items] |
|
|
| Total share-based compensation |
$ 3,134
|
$ 747
|
| X |
- DefinitionAmount of expense for award under share-based payment arrangement. Excludes amount capitalized. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.F)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_AllocatedShareBasedCompensationExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_EmployeeServiceShareBasedCompensationAllocationOfRecognizedPeriodCostsLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_PlanNameAxis=HRGN_Biostage2013EquityIncentivePlanMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_ResearchAndDevelopmentExpenseMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_SellingGeneralAndAdministrativeExpensesMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Share-based Compensation (Details Narrative) - USD ($)
|
12 Months Ended |
|
|
|
Dec. 31, 2023 |
Dec. 31, 2022 |
Dec. 29, 2023 |
May 12, 2022 |
Dec. 31, 2021 |
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
| Number of award outstanding |
3,977,289
|
2,516,924
|
|
|
2,332,603
|
| Share-based compensation arrangement by share-based payment award, options, outstanding, intrinsic value |
$ 5,728,000
|
$ 6,917,000
|
|
|
$ 294,000
|
| Share price |
|
|
|
$ 5.50
|
|
| Weighted average exercise price |
$ 4.64
|
$ 3.95
|
|
|
$ 3.93
|
| Expected dividend yield |
0.00%
|
0.00%
|
|
|
|
| Share-based compensation arrangement by share-based payment award, options, grants in period, weighted average grant date fair value |
$ 5.12
|
$ 4.23
|
|
|
|
| Performance Shares [Member] |
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
| Number of award outstanding |
773,195
|
|
|
|
|
| Unrecognized compensation expense |
$ 2,800,000
|
|
|
|
|
| Share based compensation arrangement by share based payment award unrecognized compensation costs |
$ 0
|
|
|
|
|
| HRGN Amended And Restate Equity Incentive Plan [Member] |
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
| Share-based compensation arrangement by share-based payment award, expiration period |
10 years
|
|
|
|
|
| Share-based compensation arrangement by share-based payment award, number of shares authorized |
9,098,000
|
|
|
|
|
| Number of shares available for issuance |
5,034,760
|
|
|
|
|
| Unrecognized compensation expense |
$ 3,900,000
|
|
|
|
|
| Share-based compensation arrangement by share-based payment award, options, outstanding, intrinsic value |
$ 5,700,000
|
|
|
|
|
| Share price |
|
|
$ 4.99
|
|
|
| Weighted average exercise price |
|
|
$ 4.64
|
|
|
| Employee service share-based compensation, nonvested awards, compensation cost not yet recognized, period for recognition |
2 years 2 months 12 days
|
|
|
|
|
| X |
- DefinitionAmount of share based compensation arrangement by share based payment award unrecognized compensation costs.
| Name: |
HRGN_ShareBasedCompensationArrangementByShareBasedPaymentAwardUnrecognizedCompensationCosts |
| Namespace Prefix: |
HRGN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of cost not yet recognized for nonvested award under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_EmployeeServiceShareBasedCompensationNonvestedAwardsTotalCompensationCostNotYetRecognized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted-average period over which cost not yet recognized is expected to be recognized for award under share-based payment arrangement, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_EmployeeServiceShareBasedCompensationNonvestedAwardsTotalCompensationCostNotYetRecognizedPeriodForRecognition1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe estimated dividend rate (a percentage of the share price) to be paid (expected dividends) to holders of the underlying shares over the option's term. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardFairValueAssumptionsExpectedDividendRate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares authorized for issuance under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardNumberOfSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe difference between the maximum number of shares (or other type of equity) authorized for issuance under the plan (including the effects of amendments and adjustments), and the sum of: 1) the number of shares (or other type of equity) already issued upon exercise of options or other equity-based awards under the plan; and 2) shares (or other type of equity) reserved for issuance on granting of outstanding awards, net of cancellations and forfeitures, if applicable. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardNumberOfSharesAvailableForGrant |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe weighted average grant-date fair value of options granted during the reporting period as calculated by applying the disclosed option pricing methodology. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsGrantsInPeriodWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount by which the current fair value of the underlying stock exceeds the exercise price of options outstanding. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingIntrinsicValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of options outstanding, including both vested and non-vested options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average price at which grantees can acquire the shares reserved for issuance under the stock option plan. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardOptionsOutstandingWeightedAverageExercisePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionPrice of a single share of a number of saleable stocks of a company.
| Name: |
us-gaap_SharePrice |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionPeriod from grant date that an equity-based award expires, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents the reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_SharebasedCompensationArrangementBySharebasedPaymentAwardExpirationPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_PlanNameAxis=HRGN_HRGNAmendedAndRestateEquityIncentivePlanMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
Schedule of Basic and Diluted Net Loss Per Share (Details) - USD ($)
$ / shares in Units, $ in Thousands |
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Earnings Per Share [Abstract] |
|
|
| Net loss |
$ (8,945)
|
$ (6,073)
|
| Preferred stock dividends |
(77)
|
(180)
|
| Net loss attributable to common stockholders |
$ (9,022)
|
$ (6,253)
|
| Weighted average number of shares outstanding basic |
13,455,666
|
11,349,610
|
| Weighted average number of shares outstanding diluted |
13,455,666
|
11,349,610
|
| Basic net loss per share |
$ (0.67)
|
$ (0.55)
|
| Diluted net loss per share |
$ (0.67)
|
$ (0.55)
|
| X |
- References
+ Details
| Name: |
us-gaap_EarningsPerShareAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period per each share of common stock or unit outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period available to each share of common stock or common unit outstanding during the reporting period and to each share or unit that would have been outstanding assuming the issuance of common shares or units for all dilutive potential common shares or units outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareDiluted |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, after deduction of tax, noncontrolling interests, dividends on preferred stock and participating securities; of income (loss) available to common shareholders. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 5
-Subparagraph (SAB Topic 6.B)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-5
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
Reference 11: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-11
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
| Name: |
us-gaap_NetIncomeLossAvailableToCommonStockholdersBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAccretion of temporary equity during the period due to cash, stock, and in-kind dividends. This item is an adjustment to net income necessary to derive net income apportioned to common stockholders and is to be distinguished from Temporary Equity, Accretion of Dividends (Temporary Equity, Accretion of Dividends).
| Name: |
us-gaap_TemporaryEquityDividendsAdjustment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe average number of shares or units issued and outstanding that are used in calculating diluted EPS or earnings per unit (EPU), determined based on the timing of issuance of shares or units in the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 16
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-16
| Name: |
us-gaap_WeightedAverageNumberOfDilutedSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of [basic] shares or units, after adjustment for contingently issuable shares or units and other shares or units not deemed outstanding, determined by relating the portion of time within a reporting period that common shares or units have been outstanding to the total time in that period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
| Name: |
us-gaap_WeightedAverageNumberOfSharesOutstandingBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.24.1
Schedule of Antidilutive Securities Excluded from Computation of Earnings per Share (Details) - shares
|
12 Months Ended |
Dec. 31, 2023 |
Dec. 31, 2022 |
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share, Amount |
5,090,911
|
4,249,805
|
| Warrant [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share, Amount |
1,113,622
|
1,113,622
|
| Share-Based Payment Arrangement, Option [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share, Amount |
3,977,289
|
2,516,924
|
| Series E Convertible Preferred Stock [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share, Amount |
|
|
| X |
- DefinitionSecurities (including those issuable pursuant to contingent stock agreements) that could potentially dilute basic earnings per share (EPS) or earnings per unit (EPU) in the future that were not included in the computation of diluted EPS or EPU because to do so would increase EPS or EPU amounts or decrease loss per share or unit amounts for the period presented. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=us-gaap_WarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=us-gaap_EmployeeStockOptionMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=HRGN_SeriesEConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all assets that are recognized. Assets are probable future economic benefits obtained or controlled by an entity as a result of past transactions or events. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(12))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 23: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 26: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(11))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_Assets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, excluding tax collected from customer, of revenue from satisfaction of performance obligation by transferring promised good or service to customer. Tax collected from customer is tax assessed by governmental authority that is both imposed on and concurrent with specific revenue-producing transaction, including, but not limited to, sales, use, value added and excise. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 924
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 11.L)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479941/924-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 42
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-42
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 40
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-40
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-4
| Name: |
us-gaap_RevenueFromContractWithCustomerExcludingAssessedTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_StatementBusinessSegmentsAxis=HRGN_RegenerativeBiotechMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementBusinessSegmentsAxis=HRGN_LongevityProductsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.24.1
| X |
- DefinitionAmount, before unamortized (discount) premium and debt issuance costs, of long-term debt. Includes, but is not limited to, notes payable, bonds payable, commercial loans, mortgage loans, convertible debt, subordinated debt and other types of debt. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(22))
-SubTopic 10
-Topic 210
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(16))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481139/470-20-50-4
| Name: |
us-gaap_DebtInstrumentCarryingAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionFace (par) amount of debt instrument at time of issuance. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 835
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482900/835-30-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481139/470-20-50-1B
Reference 3: http://www.xbrl.org/2003/role/exampleRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 69B
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481568/470-20-55-69B
Reference 4: http://www.xbrl.org/2003/role/exampleRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 69C
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481568/470-20-55-69C
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 835
-SubTopic 30
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482925/835-30-45-2
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 835
-SubTopic 30
-Section 55
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482949/835-30-55-8
| Name: |
us-gaap_DebtInstrumentFaceAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionThe designation of funds furnished by a borrower to a lender to assure future payments of the borrower's real estate taxes and insurance obligations with respect to a mortgaged property. Escrow deposits may be made for a variety of other purposes such as earnest money and contingent payments. This element excludes replacement reserves which are an escrow separately provided for within the US GAAP taxonomy. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 954
-SubTopic 440
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480327/954-440-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 942
-SubTopic 210
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03.10)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_EscrowDeposit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe portion of the carrying amount of short-term borrowings outstanding as of the balance sheet date which accrues interest at a set, unchanging rate.
| Name: |
us-gaap_ShortTermDebtPercentageBearingFixedInterestRate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionDetail information of subsequent event by type. User is expected to use existing line items from elsewhere in the taxonomy as the primary line items for this disclosure, which is further associated with dimension and member elements pertaining to a subsequent event. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481674/830-30-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483399/855-10-50-2
| Name: |
us-gaap_SubsequentEventLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_SubsequentEventTypeAxis=us-gaap_SubsequentEventMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_TitleOfIndividualAxis=srt_ChiefExecutiveOfficerMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
Biostage (QB) (USOTC:BSTG)
Historical Stock Chart
From Mar 2024 to Apr 2024
 Charts.")
Biostage (QB) (USOTC:BSTG)
Historical Stock Chart
From Apr 2023 to Apr 2024
 Charts.")